
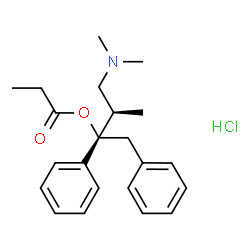
Dextropropoxyphene hydrochloride
- Molecular FormulaC22H30ClNO2
- Average mass375.932
dextropropoxyphene hydrochloride
(+)-Propoxyphene hydrochloride
(1S,2R)-1-Benzyl-3-(dimethylamino)-2-methyl-1-phenylpropylpropanoathydrochlorid [German]
(2S,3R)-4-(Dimethylamino)-3-methyl-1,2-diphenyl-2-butanyl propanoate hydrochloride (1:1) [ACD/IUPAC Name]
(2S,3R)-4-(Dimethylamino)-3-methyl-1,2-diphenyl-2-butanyl-propanoathydrochlorid (1:1) [German][ACD/IUPAC Name]
(2S,3R)-4-(Dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl propanoate hydrochloride (1:1)
(aS)-a-((1R)-2-(Dimethylamino)-1-methylethyl)-a-phenylbenzeneethanol Propanoate (Ester) Hydrochloride
1639-60-7 CAS
Algafan [Trade name]
Antalvic [Trade name]
benzeneethanol, α-[(1R)-2-(dimethylamino)-1-methylethyl]-α-phenyl-, propanoate (ester), (αS)-, hydrochloride
Dextropropoxyphene[3] is an analgesic in the opioid category, patented in 1955[4] and manufactured by Eli Lilly and Company. It is an optical isomer of levopropoxyphene. It is intended to treat mild pain and also has antitussive (cough suppressant) and local anaesthetic effects. The drug has been taken off the market in Europe and the US due to concerns of fatal overdoses and heart arrhythmias.[5] Its onset of analgesia (pain relief) is said to be 20–30 minutes and peak effects are seen about 1.5–2 hours after oral administration.[1]
Dextropropoxyphene is sometimes combined with acetaminophen. Trade names include Darvocet-N and Di-Gesic,[6] Darvon with APAP (for dextropropoxyphene and paracetamol).[7] The British approved name (i.e. the generic name of the active ingredient) of the paracetamol/dextropropoxyphene preparation is “co-proxamol” (sold under a variety of brand names); however, it has been withdrawn since 2007, and is no longer available to new patients, with exceptions.[8] The paracetamol combination(s) are known as Capadex or Di-Gesic in Australia, Lentogesic in South Africa, and Di-Antalvic in France (unlike co-proxamol, which is an approved name, these are all brand names).
Dextropropoxyphene is known under several synonyms, including:
- Alpha-d-4-dimethylamino-3-methyl-1,2-diphenyl-2-butanol propionate
- [(2S,3S)-4-(Dimethylamino)-3- methyl-1,2-diphenylbutan-2-yl] propanoate
- (+)-1,2-Diphenyl-2-propionoxy- 3-methyl-4-di-methylaminobutane
READ
www.drugfuture.com/mt/dextropropoxyphene.pdf
40 Analgesics Anti-inflammatory Drugs and Antipyretics … NOTE. Compounded preparations ofdextropropoxyphene hydro- chloride may be represented by the …

Title: Propoxyphene
CAS Registry Number: 469-62-5
CAS Name: (aS)-a-[(1R)-2-(Dimethylamino)-1-methylethyl]-a-phenylbenzeneethanol propanoate (ester)
Additional Names: a-d-4-dimethylamino-3-methyl-1,2-diphenyl-2-butanol propionate; (+)-1,2-diphenyl-2-propionoxy-3-methyl-4-dimethylaminobutane; (+)-4-dimethylamino-1,2-diphenyl-3-methyl-2-propionyloxybutane; a-d-propoxyphene; dextropropoxyphene
Molecular Formula: C22H29NO2
Molecular Weight: 339.47
Percent Composition: C 77.84%, H 8.61%, N 4.13%, O 9.43%
Literature References: Prepn of racemate: Pohland, Sullivan, J. Am. Chem. Soc. 75, 4458 (1953); Pohland, US 2728779 (1955 to Lilly). Prepn of (+)-form: Pohland, Sullivan, J. Am. Chem. Soc. 77, 3400 (1955). Stereochemistry: Sullivan et al., J. Org. Chem.28, 2381 (1963); Casy, Myers, J. Pharm. Pharmacol. 16, 455 (1964). Stereospecific synthesis: Pohland et al., J. Org. Chem. 28,2483 (1963). Metabolism: S. L. Due et al., Biomed. Mass Spectrom. 3, 217 (1976). The a-dl- and d-diastereoisomers possess marked analgesic activity in contrast to the b-diastereoisomers which are substantially inactive. Toxicity: E. I. Goldenthal, Toxicol. Appl. Pharmacol. 18, 185 (1971); J. L. Emerson et al., ibid. 19, 445 (1971). Comprehensive description: B. McEwan, Anal. Profiles Drug Subs. 1, 301-318 (1972). Symposium on pharmacology, toxicology, and clinical efficacy of propoxyphene alone and in combination with acetaminophen: Hum. Toxicol. 3, Suppl., 1S-238S (1984).
Properties: Crystals from petr ether, mp 75-76°. [a]D25 +67.3° (c = 0.6 in chloroform).
Melting point: mp 75-76°
Optical Rotation: [a]D25 +67.3° (c = 0.6 in chloroform)
Derivative Type: Hydrochloride
CAS Registry Number: 1639-60-7
Trademarks: Darvon (AAI Pharma); Deprancol (Parke-Davis); Develin (Gecke)
Molecular Formula: C22H29NO2.HCl
Molecular Weight: 375.93
Percent Composition: C 70.29%, H 8.04%, N 3.73%, O 8.51%, Cl 9.43%
Properties: Bitter crystals from methanol + ethyl acetate, mp 163-168.5°. [a]D25 +59.8° (c = 0.6 in water). Sol in water, alc, chloroform, acetone. Practically insol in benzene, ether. LD50 in mice, rats (mg/kg): 28, 15 i.v.; 111, 58 i.p.; 211, 134 s.c.; 282, 230 orally (Emerson).
Melting point: mp 163-168.5°
Optical Rotation: [a]D25 +59.8° (c = 0.6 in water)
Toxicity data: LD50 in mice, rats (mg/kg): 28, 15 i.v.; 111, 58 i.p.; 211, 134 s.c.; 282, 230 orally (Emerson)
Derivative Type: Napsylate monohydrate
CAS Registry Number: 26570-10-5
Trademarks: Darvon-N (AAI Pharma)
Molecular Formula: C22H29NO2.C10H8O3S.H2O
Molecular Weight: 565.72
Percent Composition: C 67.94%, H 6.95%, N 2.48%, O 16.97%, S 5.67%
Properties: Odorless, white crystalline powder; bitter taste. Sol in methanol, ethanol, chloroform, acetone; very slightly sol in water. LD50 orally in female rats: 990 mg/kg (Goldenthal).
Toxicity data: Odorless, white crystalline powder; bitter taste. Sol in methanol, ethanol, chloroform, acetone; very slightly sol in water. LD50 orally in female rats: 990 mg/kg (Goldenthal)
Derivative Type: a-l-Form see Levopropoxyphene
Derivative Type: a-dl-Form
Additional Names: Racemic propoxyphene; diméprotane
Derivative Type: a-dl-Form hydrochloride
Properties: Crystals from methanol + ethyl acetate, mp 170-171°. Soluble in water, alcohol, chloroform. Practically insol in benzene, ether.
Melting point: mp 170-171°
Derivative Type: b-dl-Form
Properties: Crystals from acetone + ether. mp 187-188°. More soluble than the a-form.
Melting point: mp 187-188°
NOTE: Bulk dextropropoxyphene (non-dosage forms) is a controlled substance (opiate): 21 CFR, 1308.12; dextropropoxyphene is a controlled substance (narcotic): 21 CFR, 1308.14.
Therap-Cat: Analgesic (narcotic).
Keywords: Analgesic (Narcotic).
Of the many phenylpropylamines which show analgesic activity, the two most important are methadone and propoxyphene. The optically active alpha-dextro 0 stereoisomer of propoxyphene is the only stereoisomer of propoxyphene which possesses analgesic properties. It is commonly used in its hydrochloride salt form which is a bitter, white crystalline powder freely soluble in water and soluble in alcohol. Its chemical name is $ eC-d-1 ,2-diphenyl-2-propionoxy-3-methyl-4-dimethyl- aminobutane hydrochloride and is sold under several different trademarks including, for example, DARVON, DOLENE, and SK-65. The napsylate salt, i.e. , the naphthalene sulfonate, is also used in many drug forms. 0 It has previously been made from the hydrochloride salt.
Preparation of d-propoxyphene hydrochloride was first described by A. Pohland and H.R. Sullivan at J. Am. Chem. Soc. , Volume 75, pp. 4458(1953). Therein, the authors disclosed a synthesis involving several stages, (1) preparation of an aminoketone called
,-dimethylaminobutrophenone by addition of the secondary amine to phenylpropenyl ketone; (2) a Grignard reaction of the amino ketone with benzylmagnesium chloride to yield the amino, hydrochloride-carbinols described as Λr(75%) and ^-(15%) 4-dimethylamino-l ,2-diphenyl-3-methyl-2-butanol hydrochloride (sometimes hereinafter referred to as d-oxyphene hydrochloride); and (3) acylation of the flCramino carbinol hydrochloride by addition of an equal weight of propionic anhydride and five times that weight of pyridine and heating- to reflux for several hours. Note the following reaction formula:

After cooling to recover the crude product, it was purified by two recrystallizations from methanol-ethyl acetate solution resulting in a yield of 70%.
Although this work confirmed that the oc and not the -diastereoisomers of propoxyphene gave rise to analgesic activity, it was still necessary to determine which of the optical forms of the ■aC-diastereoisomer, i.e. βC-d(+) or ύL-l(-), was responsible for the analgesic activity. Accordingly, Pohland and Sullivan reported in the J. Am. Chem. Soc. , Volume 77, pp. 3400 (1955) their work on resolution of α.-dl-4-dimethylamino-l , 2-diphenyl-3-methyl-2-butanol by fractional crystallization of its d-camphorsulfonic acid salt. From the respective d-d and α-1 carbinol d-camphor- sulfonic salts the optically active hydrochloride salts were prepared. The ofc-d-hydrochloride was acylated using propionic anhydride and triethylamine, while the ot-1 hydrochloride was acylated using propionic anhydride and pyridine. It was therein found that only the C-d stereoisomer gave the analgesic response. However, final purification of the hydrochloride salt required additional HC1 and three recrystallizations and with yields of less than about 70%.
In 1963, Pohland, Peters and Sullivan reported in the J. Org. Chem. , Vol. 28, pp. 2483, an alternative synthetic route for Λ-d-propoxyphene hydrochloride. Working backwards from the desired optically active isomer of propoxyphene by its hydrolysis and dehydration to stilbene, followed by ozonization of the stilbene, the authors discovered good yield of (-)-^-dimethyl- amino-^rmethylpropiophenone. This optically active amino ketone was found to be surprisingly stable in salt form thus permitting its use as a starting material for a stereo selective synthesis of ύC-d-propoxyphene. Racemic ^-dimethylamino-tf-methylpropiophenone was resolved by crystallization of the dibenzoyl tartrate salts from acetone solution. The use of dibenzoyl-(-) -tartaric acid yielded the insoluble salt having (-)-^-dimethylamino-<-methylpropiophenone, while the use of the (+) tartaric acid yielded the salt having the (+) amino ketone isomer.
It is of interest that according to this reported synthesis, it was the (-) isomer of/6-dimethylamino— efc-methylpropiophenone, which when liberated from its (-) tartrate salt by Grignard reaction with benzylmagnesium chloride provided good yields of the (+) or (d) isomer ύfc-1,2-diphenyl-3-methylτ4-dimethylamino-2-butanol which of course is the carbinol precursor for oC-d-propoxyphene. The reported yields were 69%. The acylation was accomplished as had been previously reported, i.e., by means of propionic anhydride in either triethylamine or pyridine.
In 1978, Hungarian Pat. No. 14,441 disclosed a synthesis of cfc-d-propoxyphene employing the above-described method except that (1) the (+) tartaric acid was employed in the resolution of the racemic ^-dimethylamino-ot-methylpropiophenone and (2) the acylation was accomplished by reacting triethylamine in chloroform, propionyl chloride and the carbinol rather than propionyl anhydride and the carbinol hydrochloride. Still the product was precipitated in ether and required an amine catalyst.
Most recently, U.S. Patent Number 4,661,625 disclosed a synthetic method involving acylation of the carbinol (d-oxyphene) with propionyl chloride and thionyl chloride in dichloromethane. The yield of d-propoxyphene hydrochloride was improved to at least 76%, but use of the toxic additive thionyl chloride was required to get to that level. In addition, methylene chloride or another chlorinated solvent was required. Chlorinated impurities resulted and caused difficulties in purification.
However, a method that provides even higher yields of d-propoxyphene and its salts and doesn’t require toxic and/or hazardous additives and solvents has long been highly desired. It is an object of the present invention to provide a means of producing d-propoxyphene in high yields without the need for amines or chlorinated solvents. It is a further object to provide methods of producing the hydrochloride and napsylate salts of d-propoxyphene in higher yields than previously obtainable.
Patent
https://www.google.com/patents/WO1990014331A1?cl=en
Example 1
To a 5-litre flask equipped with an overhead stirrer, a nitrogen feed, thermometer, and heating mantle was added 2.0 kg (7.06 moles) d-oxyphene purchased from Merrell-Dow. To this was added 2.0 L (15.6 moles) propionic anhydride (Aldrich) with stirring and heating. The temperature was raised to 75-80 °C over 35 minutes and maintained at no more than 81 °C for four hours. The mixture was cooled to room temperature and then added dropwise to 10.0 L deionized water over 30 minutes. A clear yellow solution resulted. 1.85 L ammonium hydroxide was added to raise the pH to 8.8. Seed crystals of d-propoxyphene were added and white solids precipitated. The solution and precipitate were chilled by immersion in ice bath and filtered. The solid was dried by vacuum and then placed in a 60 βC oven for 2 days. The yield was 2390 g of white crystals, 99.7% of theory.
Example 2
To a 22-litre vessel equipped with a stirrer. heating mantle, thermometer and a nitrogen feed was added 5.0 kg (17.6) moles d-oxyphene (Merrell-Dow). To it was added 5.0 L (39.0 moles) propionic anhydride (Eastman-Kodak). The temperature was raised and varied from 73-88 βC over 4 1/4 hours. The reaction mixture was split into two parts, each about 5L, and each was treated as follows: The mixture was slowly added to a mixture of 3.125 L absolute ethanol and 9.37 L deionized water. A mild odor of ethyl propionate was noted, but no phase separation was seen. The pH was adjusted to 8.8 with ammonium hydroxide, and white solids slowly precipitated. The mixture was chilled to approximately 9 °C and filtered with vacuum. The solids were washed twice on the filter with 2 L deionized water and reslurried in 10 L at room temperature. They were refiltered and again washed twice with 2 L deionized water. The solids were air-dried and analyzed by NMR, which showed no ethanol remaining. The combined yield was 6.12 kg, 102% of theory, m.p. 73.8 – 75.1 βC.
Example 3
A 100-g sample of d-propoxyphene prepared as in Example 1 was dissolved in 481 mL ethyl acetate. 26.0 mL methanolic HC1 (11.7 M) was added. The mixture was warmed to between 30 and 40 °C. The mixture then slowly crystallized. It was cooled to below 5 °C and filtered. The crystals were washed with 50 mL cold ethyl acetate. The yield was 79.3 g (72%).
Unconverted free base was recovered from the ethyl acetate filtrate by twice extracting with 100 mL of water acidified with 5 drops cone. HCl. The aqueous extracts were combined, and the remaining ethyl acetate was removed by blowing air over the solution. When the ethyl acetate was completely removed, the pH was raised to 9.0 by adding ammonium hydroxide; solids formed that were filtered, washed and dried. 25.3 g d-propoxyphene were recovered. The total yield of salt and recovered free base was 94.5%.
Example 4
A 40-g sample of d-propoxyphene prepared as in Example 2 was slurried in 169 mL deionized water with stirring and 10.9 mL cone. HC1 were added.
Seventy mL ethanol were added; then 30.15 g sodium napsylate were added with stirring. The resulting slurry was heated to between 50 and 60 °C until a solution was obtained. The solution was filtered while hot and then allowed to cool, with stirring. Crystallization started on cooling. The solution was then chilled to less than 5 °C and filtered. The solids were washed with 210 mL deionized water and then reslurried in 195 mL deionized water. The slurry was stirred for 15 minutes, then filtered. The solids were again washed with 210 mL deionized water, collected, and dried overnight at 50-60 °C. The yield was 63.6 g (95.4 percent of theory).
PATENT

Patent EP0225778B1 – Improved synthesis and purification of alpha …
References
- ^ Jump up to:a b c d e Davis, MP; Glare, PA; Hardy, J (2009) [2005]. Opioids in Cancer Pain (2nd ed.). Oxford, UK: Oxford University Press. ISBN 978-0-19-157532-7.
- Jump up^ “PRODUCT INFORMATION PARADEX” (PDF). TGA eBusiness Services. Aspen Pharmacare Australia Pty Ltd. 2 March 2010. Retrieved 9 April 2014.
- Jump up^ US Patent 2728779 – Esters of Substituted Aminobutanes
- Jump up^ Thieme Chemistry (Hrsg.): RÖMPP Online – Version 3.32. Georg Thieme Verlag KG, Stuttgart, 30. April 2013.
- Jump up^ Physicians Say Good Riddance to ‘Worst Drug in History’ By Allison Gandey. February 2, 2011
- Jump up^ “Consumer Medicine Information: Digesic” (PDF). Aspen Pharmacare Australia Pty Ltd.
- Jump up^ Nursing Drug Handbook, Springhouse, page 306
- Jump up^ BNF Edition 57, BNF.org
- Jump up^ “Restless legs syndrome: Definition from”. Answers.com. Retrieved 2009-08-19.
- Jump up^ “Restless Leg Syndrome – Sleep Medicine Centers of WNY”. Sleepmedicinecenters.com. Retrieved 2009-08-19.
- Jump up^ “Causes, diagnosis and treatment for the patient living with Restless Legs Syndrome (RLS)”. Restless Leg Syndrome Foundation. 1 April 2006. Retrieved 2009-08-19.
- Jump up^ http://www.aspenpharma.com.au/product_info/pi/PI_Sigma-Dexamphetamine.pdf
- Jump up^ “Blockade of Rat α3β4 Nicotinic Receptor Function by Methadone, Its Metabolites, and Structural Analogs — JPET”.
- ^ Jump up to:a b c Nickander et al., 1984
- ^ Jump up to:a b c Strom et al., 1985b
- Jump up^ Holland & Steinberg, 1979
- Jump up^ Bredgaard, Sorensen et al., 1984
- Jump up^ Stork et al., 1995
- Jump up^ Wilson, Charles Owens; Gisvold, John H. Wilson and Gisvold’s textbook of organic medicinal and pharmaceutical chemistry. Lippincott Williams & Wilkins. ISBN 0-7817-3481-9.
- Jump up^ “FDA pulls common pain med off the market”. November 19, 2010. CNN.
- Jump up^ http://www.tga.gov.au/newsroom/media-2011-dextropropoxyphene-111122.htm
- Jump up^ “Legal win keeps banned painkiller on the shelves”. The Sydney Morning Herald. 22 February 2012.
- Jump up^ “Questions and answers on the withdrawal of the marketing authorisations for medicines containing dextropropoxyphene” (PDF). European Medicines Agency. 25 June 2009. Retrieved 2009-09-08.
- Jump up^ “Paradex And Capadex To Be Withdrawn From NZ”. Retrieved 2010-02-21.
- Jump up^ “Fasta kombinationer av smärtstillande läkemedel innehållande dextropropoxifen försvinner från marknaden under hösten 2005” [Fixed combinations of analgesic drugs containing dextropropoxyphene disappear from the market in the autumn of 2005] (in Swedish). Läkemedelsverket. 5 May 2005.
- Jump up^ EU-decision http://www.medicinesauthority.gov.mt/pub/Dextropoxyphene.pdf
- Jump up^ “Dextropropoxyphene is removed from the market” (in Swedish). 5 May 2005.
- Jump up^ Painkiller Scrapped Over Suicides
- Jump up^ “Distalgesic (discontinued in the UK – December 2007)”. netdoctor. 9 January 2008. Retrieved 8 September 2013.
- Jump up^ BNF.org, BNF edition 57, Retrieved 28 August 2009
- ^ Jump up to:a b “Withdrawal of co-proxamol products and interim updated prescribing information”.Medicines and Healthcare products Regulatory Agency (MHRA). 31 January 2005. Retrieved August 28, 2009.
- Jump up^ “Outcome of the public request for information on the risks and benefits of the pain killer co-proxamol”. Medicines and Healthcare products Regulatory Agency (MHRA).
- Jump up^ “Co-proxamol: outcome of the review of risks and benefits”. Questions and Answers leaflet, Retrieved August 28, 2009
- Jump up^ Hawton K, Bergen H, Simkin S, Brock A, Griffiths C, Romeri E, Smith KL, Kapur N, Gunnell D (June 2009). “Effect of withdrawal of co-proxamol on prescribing and deaths from drug poisoning in England and Wales: time series analysis” (PDF). BMJ. 338: b2270. doi:10.1136/bmj.b2270. PMC 3269903
 . PMID 19541707.
. PMID 19541707.
- Jump up^ Co-Proxamol: 13 Jul 2005: House of Commons debates (TheyWorkForYou.com)
- Jump up^ Co-proxamol: 17 Jan 2007: Westminster Hall debates (TheyWorkForYou.com)
- Jump up^ Failure Of MHRA Coproxamol Named Patient System – Visitor Opinion
- Jump up^ News Centre : MHRA
- Jump up^ “FDA Takes Actions on Darvon, Other Pain Medications Containing Propoxyphene”.U.S. Food and Drug Administration (FDA). 7 July 2009.
- Jump up^ Drug Information for Darvocet-N 100 Oral – Web MD
- Jump up^ NCQA’s HEDIS Measure: Use of High Risk Medications in the Elderly
- Jump up^ Zajac, Andrew (November 19, 2010). “Darvon, Darvocet painkillers pulled from the U.S. market”. L.A. Times. Archived from the original on November 22, 2010. RetrievedNovember 19, 2010.
- Jump up^ Health Canada: Darvon-N (dextropopoxyphene) – Recall and Withdrawal in Canada
- Jump up^ The Hindu: Govt. bans painkiller
- ^ Jump up to:a b Nitschke, Philip; Fiona Stewart (2006). The Peaceful Pill Handbook. U.S.: Exit International. ISBN 0-9788788-1-7.
- Jump up^ Pieter Admiraal; et al. Guide to a Humane Self-Chosen Death. The Netherlands: WOZZ Foundation, Delft. ISBN 90-78581-01-8.
- Jump up^ ASH Wiki: Darvon Cocktail
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| US4661625 * |
Dec 2, 1985 |
Apr 28, 1987 |
Mallinckkodt, Inc. |
Synthesis and purification of d-propoxyphene hydrochloride |
///////////
CCC(=O)OC(CC1=CC=CC=C1)(C2=CC=CC=C2)C(C)CN(C)C








 The presentation will load below
The presentation will load below CHEMTRIX
CHEMTRIX


























 Amerikansk lovgivning holder hånden under Novozymes, når der tales om majsbaseret bioethanol, da det er lovfæstet, at 10 pct. af brændstofforbruget skal kommer fra biobrændstof
Amerikansk lovgivning holder hånden under Novozymes, når der tales om majsbaseret bioethanol, da det er lovfæstet, at 10 pct. af brændstofforbruget skal kommer fra biobrændstof

