Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Pronay Das†ab, Palak Babbar†c, Nipun Malhotra†c, Manmohan Sharmac , Goraknath R. Jachakab , Rajesh G. Gonnadebd, Dhanasekaran Shanmugambe, Karl Harlosf , Manickam Yogavelc , Amit Sharmac *, and D. Srinivasa Reddyab* †All three have contributed equally to this work.
aOrganic Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
b Academy of Scientific and Innovative Research (AcSIR), New Delhi 110025, India
cMolecular Medicine Group, International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi 110067, India dCenter for Material Characterization, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
e Biochemical Sciences Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
fDivision of Structural Biology, Welcome Trust Centre for Human Genetics, The Nuffield Department of Medicine, University of Oxford, Oxford OX3 7BN, UK
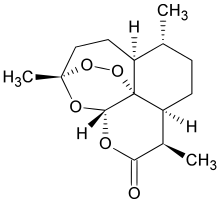
The dependence of drug potency on diastereomeric configurations is a key facet. Using a novel general divergent synthetic route for a three-chiral centre anti-malarial natural product cladosporin, we built its complete library of stereoisomers (cladologs) and assessed their inhibitory potential using parasite-, enzyme- and structure-based assays.
We show that potency is manifest via tetrahyropyran ring conformations that are housed in the ribose binding pocket of parasite lysyl tRNA synthetase (KRS). Strikingly, drug potency between top and worst enantiomers varied 500-fold, and structures of KRS-cladolog complexes reveal that alterations at C3 and C10 are detrimental to drug potency where changes at C3 are sensed by rotameric flipping of Glutamate332.
Given that scores of anti-malarial and anti-infective drugs contain chiral centers, this work provides a new foundation for focusing on inhibitor stereochemistry as a facet of anti-microbial drug development.
Cladosporin (12) displays exquisite selectivity for the parasite lysyl-tRNA synthetase over human enzyme. This species specific selectivity of cladosporin has been previously described through comprehensive sequence alignment, where the residues val329 and ser346 seem to be sterically crucial for accommodating the methyl moiety of THP ring10. The structural features of compound 12 clearly indicate the presence of three stereocenters, and therefore 2n (n=3) i.e., eight stereoisomers are possible (Fig.1). Till date, only one asymmetric total synthesis of cladosporin13 has been achieved which was followed by another report of formal syntheses14. Here, we have developed a general chemical synthesis route to synthetically access all the eight possible stereoisomers of compound 12.
The research interests of his group lie in issues related to application of oriented organic synthesis, in particular total synthesis of biologically active natural products, medicinal chemistry and crop protection. This team has been credited with having accomplished total synthesis of more than 25 natural products with impressive biological activities. “Some of our recent achievements include identification of potential leads, like antibiotic compound based on hunanamycin natural product for treating food infections, anti-diabetic molecule in collaboration with an industry partner and anti-TB compound using a strategy called ‘re-purposing of a drug scaffold’,” said Reddy.
A total of two awardees out of four were from CSIR institutes. In addition to Reddy, Rajan Shankarnarayanan, CSIR – CCMB, Hyderabad (basic sciences), also was conferred with the award. Vikram Mathews, CMC, Vellore (medical research) and Prof Ashish Suri, AIIMS, New Delhi (clinical research), were the others to receive the awards.
With more than 80 scientific publications and 35 patents, Reddy is one of the most prominent scientists in the city and has already been honoured with the Shanti Swarup Bhatnagar prize in chemical sciences. Reddy is also a nominated member of the scientific body of Indian Pharmacopoeia, government of India and was elected as a fellow of the Telangana and Maharashtra Academies of Sciences in addition to the National Academy of Sciences, India (NASI).
Merck Serono, the biopharmaceutical business of Merck, and MMV announced today that an agreement has been signed for Merck Serono to obtain the rights to the investigational antimalarial compound DDD107498 from MMV. This agreement underscores the commitment of Merck Serono to provide antimalarials for the most vulnerable populations in need.
“This agreement strengthens our Global Health research program and our ongoing collaboration with Medicines for Malaria Venture,” said Luciano Rossetti, Executive Vice President, Global Head of Research & Development at Merck Serono. “MMV is known worldwide for its major contribution to delivering innovative antimalarial treatments to the most vulnerable populations suffering from this disease, and at Merck Serono we share this goal.”
DDD107498 originated from a collaboration between MMV and the University of Dundee Drug Discovery Unit, led by Prof. Ian Gilbert and Dr. Kevin Read. The objective of the clinical program is to demonstrate whether the investigational compound exerts activity on a number of malaria parasite lifecycle stages, and remains active in the body long enough to offer potential as a single-dose treatment against the most severe strains of malaria.
While development and commercialization of the compound is under Merck Serono’s responsibility, MMV will provide expertise in the field of malaria drug development, including its clinical and delivery expertise, and provide access to its public and private sector networks in malaria-endemic countries.
Merck Serono has a dedicated Global Health R&D group working to address key unmet medical needs related to neglected diseases, such as schistosomiasis and malaria, with a focus on pediatric populations in developing countries. Its approach is based on public-private partnerships and collaborations with leading global health institutions and organizations in both developed and developing countries.
“Working with partners like Merck Serono is critical to the progress of potential antimalarial compounds, like DDD107498, through the malaria drug pipeline,” said Dr. Timothy Wells, Chief Scientific Officer at MMV. “Their Global Health Program is gaining momentum and we need more compounds to tackle malaria, a disease that places a huge burden on the world’s most vulnerable populations. DDD107498 holds great promise and we look forward to working with the Merck Serono team through the development phase.”
According to the World Health Organization, there were an estimated 198 million cases of malaria worldwide in 2013, and an estimated 584,000 deaths, primarily in young children from the developing world. The launch of the not-for-profit research foundation, MMV, in 1999 and a number of collaborations and partnerships, including those with Merck Serono, has contributed to reducing the major gap in malaria R&D investment and subsequent dearth of new medicines.
“It’s hugely encouraging to see the German pharmaceutical industry increasing their engagement in the development of novel antimalarials,” said global malaria expert Prof. Dr. Peter Kremsner, Director of the Institute for Tropical Medicine at the University of Tübingen, Germany. “The Merck Serono and MMV collaboration to develop DDD107498 is a great step. It’s a compound that offers lots of promise so I’m excited to see how it progresses.
Scots scientists in ‘single dose’ malaria treatment breakthrough
STV
An antimalarial drug that could treat patients was discovered by Dundee university scientists
Scientists have discovered an antimalarial compound that could treat malaria patients in a single dose and help prevent the spread of the disease from infected people.
The compound DDD107498 also has the potential to treat patients with malaria parasites resistant to current medications, researchers say.
Scientists hope it could lead to treatments and protection against the disease, which claimed almost 600,000 lives amid 200 million reported cases in 2013.
The compound was identified through a collaboration between the University of Dundee’s drug discovery unit (DDU) and the Medicines for Malaria Venture (MMV), a separate organisation.
The compound is now undergoing further safety testing with a view to entering human clinical trials within the next year.
Details of the discovery have been published in the journal Nature.
Professor Ian Gilbert, head of chemistry at the DDU, who led the team that discovered the compound, said: “The publication describes the discovery and profiling of this exciting new compound.
“It reveals that DDD107498 has the potential to treat malaria with a single dose, prevent the spread of malaria from infected people and protect a person from developing the disease in the first place.
“There is still some way to go before the compound can be given to patients. However, we are very excited by the progress that we have made.”
The World Health Organisation reports that there were 200 million clinical cases of malaria in 2013, with 584,000 people dying from the disease. Most of these deaths were children under the age of five and pregnant women.
MMV chief executive officer Dr David Reddy said: “Malaria continues to threaten almost half of the world’s population – the half that can least afford it.
“DDD107498 is an exciting compound since it holds the promise to not only treat but also protect these vulnerable populations.
“The collaboration to identify and progress the compound, led by the drug discovery unit at the University of Dundee, drew on MMV’s network of scientists from Melbourne to San Diego.”The publication of the research is an important step and a clear testament to the power of partnership.”
MMV selected DDD107498 to enter preclinical development in October 2013 following the recommendation of its expert scientific advisory committee.
Since then, with MMV’s leadership, large quantities of the compound have been produced and it is undergoing further safety testing with a view to entering human clinical trials within the next year.
Merck Serono has recently obtained the right to develop and, if successful, commercialise the compound, with the input of MMV’s expertise in the field of malaria drug development and access and delivery in malaria-endemic countries.
Dr Michael Chew from the Wellcome Trust, which provides funding for the DDU and MMV, said: “The need for new antimalarial drugs is more urgent than ever before, with emerging strains of the parasite now showing resistance against the best available drugs.
“These strains are already present at the Myanmar-Indian border and it’s a race against time to stop resistance spreading to the most vulnerable populations in Africa.
“The discovery of this new antimalarial agent, which has shown remarkable potency against multiple stages of the malaria lifecycle, is an exciting prospect in the hunt for viable new treatments.”
PAPER
Discovery of a Quinoline-4-carboxamide Derivative with a Novel Mechanism of Action, Multistage Antimalarial Activity, and Potent in Vivo Efficacy
Conditions: (a) morpholine, Et3N, DCM, 16 h, 72% yield; (b) MeMgBr, toluene, reflux, 4 h and then a 10% aqueous HCl, reflux, 1 h, 70% yield; (c) NBS, benzoyl peroxide, dichlorobenzene, 140 °C, 16 h, 70% yield; (d) morpholine, K2CO3, acetonitrile, 40 °C, 16 h, 64% yield; (e) 5-fluoroisatin, KOH, EtOH, 120 °C, microwave, 20 min, 30–76% yield; (f) amine, CDMT, N-methylmorpholine, DCM, 20–61% yield.
A single-dose treatment against malaria worked in mice to cure them of the disease. The drug also worked to block infection in healthy mice and to stop transmission, according to a study published in Nature today. The fact that the drug can act against so many stages of malaria is pretty new, but what’s even more exciting is the compound’s mode of action: it kills malaria in a completely new way, researchers say. The feature would make it a welcome addition to our roster of antimalarials — a roster that’s threatened by drug resistance.
RESEARCHERS SIFTED THROUGH A LIBRARY OF ABOUT 4,700 COMPOUNDS TO FIND THIS ONE
Malaria is an infectious disease that’s transmitted through mosquito bites; it’s also a leading cause of death in a number of developing countries. Approximately 3.4 billion people live in areas where malaria poses a real threat. As a result, there were 207 million cases of malaria in 2012 — and 627,000 deaths. There are drugs that can be used to prevent malaria, and even treat it, but drug resistance is halting the use of certain treatments in some areas.
A long search
Searching for a new drug is all about trial and error. To find this particular compound, researchers sifted through a library of about 4,700 compounds, testing them to see if they were capable of killing the malaria parasite in a lab setting. When they found something that worked, they tweaked the drug candidate to see if it could perform more effectively. “We went through a lot of these cycles of testing and designing new compounds,” says Ian Gilbert, a medicinal chemist at the University of Dundee in the UK, and a co-author of the study. “Eventually we optimized to the compound which is the subject of the paper.” For now, that compound’s unwieldy name is DDD107498.
To make sure DDD107498 really had potential, the researchers tested it on mice that had already been infected with malaria. A single dose was enough to provoke a 90 percent reduction in the number of parasites in their blood. The scientists also gave the compound to healthy mice that were subsequently exposed to malaria. DDD107498 helped the mice evade infection with a single dose, but it’s unclear how long that effect would last in humans. Finally, the researchers looked at whether the compound could prevent the transmission from an infected mouse to a mosquito. A day after receiving the treatment, mice were put in contact with mosquitoes. The scientists noted a 91 percent reduction in infected mosquitoes.
“IT HAS THE ABILITY TO BE A ONE-DOSE [DRUG], IN COMBINATION WITH ANOTHER MOLECULE.”
“What’s exciting about this molecule is obviously the fact that it has the ability to be a one-dose [drug], in combination with another molecule to cure blood stage malaria,” says Kevin Read, a drug researcher also at the University of Dundee and a co-author of the study. The fact that the compound has the ability to block transmission and protect against infection is equally thrilling. But the way in which DDD107498 kills malaria might be its most interesting feature. It halts the production of proteins — which are necessary for the parasite’s survival. No other malaria drug does that right now, Read says. “So, in principle, there’s no resistance out there already to this mechanism.”
The drug hasn’t been tested in humans yet, so it may not be nearly as good in the field. But Read says DDD107498 looks promising. “From all the pre-clinical or non-clinical data we’ve generated, it is comparable or better than any of the current marketed anti-malarials in those studies.” And at $1 per treatment, the price of the drug should fall “within the range of what’s acceptable,” he says.
“It looks like an excellent study, and the results look very important,” says Philip Rosenthal, a malaria drug researcher at The University of California-San Francisco who didn’t participate in the study. This is a big shift for Rosenthal’s field. Five years ago, “we had very little going on in anti-malarial drug discovery,” he says. Now, there’s quite a bit going on for malaria researchers, and a number of promising compounds are moving along. DDD107498 “is another player, and it’s got a number of positive features,” he says.
OTHER TREATMENTS HAVE TO BE TAKEN FOR A FEW DAYS
One of the features is the drug’s potency. It’s very active against cultured malaria parasites, Rosenthal says. But what’s perhaps most intriguing about DDD107498 is that the drug works against the mechanism that enables protein synthesis the malaria parasite’s cells. No other malaria drug does that right now, Read says. “Considering challenges of treating malaria, which is often in rural areas and developing countries, a single dose would be a big plus,” he says. “In addition, because of it’s long half life, it may also work to prevent malaria with once a week dosing, which is also a benefit.”
Still, no drug is perfect. The data suggests that DDD107498 doesn’t kill malaria as quickly as some other drugs, Rosenthal says. And when the researchers tested it to see how long it might take for resistance to develop, the results weren’t as promising as he would like. The parasites figured out a way to become resistant to the compound “relatively easily,” he says. That shouldn’t be “deal-killer,” however. “Its slow onset of action probably means it should be combined with a faster-acting drug,” he says.
BUT IT’S SLOW-ACTING
The compound is going through safety testing now. If everything goes well, it should hit human trials within the next year, Read says. Chances are, it will have to be used in combination with other malaria drugs, Gilbert says. “All anti-malarials are given in combination because it slows down resistance.”
“When you’re treating infectious diseases, you know that drug resistance is always a potential problem, so having a number of choices to treat malaria is a good thing,” Rosenthal says. In this case, the drug’s new mode of action may hold lead to an entirely new weapon against malaria. “Obviously it’s got a long way to go,” Read says. But the compound is “very exciting,” nonetheless.
Example 16-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1 in Scheme 2
In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4-(morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130° C. under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHCO3 saturated aqueous solution (2×100 ml). The organic layer was separated, dried over MgSO4 and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10% B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60-200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50° C. for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
Example 26-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2
The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
Example 1AAlternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4
To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHCO3 saturated aqueous solution (2×100 ml) and the organic phase was separated, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10% B and then 15 min hold at 10% B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23% B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
Example 1 : 6-Fluoro-2-r4-(morpholinomethyl)phenyll-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide, Example compound 1 in Scheme 2
In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1- ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4- (morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130°C under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHC03 saturated aqueous solution (2 x 100 ml). The organic layer was separated, dried over MgS04and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10 % B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60- 200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50°C for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
Example 2: 6-Fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2
The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
Example 1A: Alternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2- pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4
To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro- 4,6-dimethoxy-1 ,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHC03 saturated aqueous solution (2x 100 ml) and the organic phase was separated, dried over MgS04 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10 %B and then 15 min hold at 10%B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23 % B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
A Quinoline Carboxamide Antimalarial Drug Candidate Uniquely Targets Plasmodia at Three Stages of the Parasite Life Cycle
Angewandte Chemie, International Edition (2015), 54, (46), 13504-13506
Putting a stop to malaria: Phenotypic screening against malaria parasites, hit identification, and efficient lead optimization have delivered the preclinical candidate antimalarial DDD107498. This molecule is distinctive in that it has potential for use as a single-dose cure for malaria and shows a unique broad spectrum of activity against the liver, blood, and mosquito stages of the parasite life cycle.
Prof. P. M. O’Neill Department of Chemistry, University of Liverpool Liverpool, L69 7ZD (UK) E-mail: [email protected] Prof. S. A. Ward Liverpool School of Tropical Medicine, Pembroke Place Liverpool, L3 5QA (UK)
Professor Ian Gilbert FRSC
Design and synthesis of potential therapeutic agents
Position:
Professor of Medicinal Chemistry and Head of the Division of Biological Chemistry and Drug Discovery
I am a medicinal chemist and my research interests are in the design and synthesis of potential drugs. The mainstay of my work is synthetic medicinal chemistry as part of the Drug Discovery Unit (DDU). Where possible we make extensive use of molecular modeling to guide our synthetic efforts. I have a particular interests in the following aspects of drug discovery:
Neglected diseases such as human African trypanosomiasis, leishmaniasis and malaria.
Chemical validation of drug targets, including novel targets for which there is little or no precedence for drug discovery.
Novel approaches to and paradigms for drug discovery.
Mode of action studies and target identification.
I am Head of Chemistry in the DDU. Our main focuses are on neglected diseases and novel drug targets. The neglected diseases we are tackling are malaria, tuberculosis and the kinetoplastid diseases. We use both target-based approaches and phenotypic approaches (whole parasite screening). We have had particular success in validating the enzyme N-myristoyltransferase as a drug target in human African trypanosomiasis, and in identifying and optimising phenotypic hits. In our novel targets area, we aim to validate novel areas of biology as potential drug targets.
Novel process for preparing artemisinin or its derivatives such as dihydroartemisinin, artemether, arteether and artesunate. Also claims novel intermediates of artemesinin such as artemisinic acid or dihydroartemisinic acid. Discloses the use of artemisinin or its derivatives, for treating malaria, cancer, viral and parasitic infections.
In July 2016, Newport Premium™ reported that IPCA was capable of producing commercial quantities of artemether, arteether and artesunate; and holds an inactive US DMF for artemether since February 2009. In July 2016, IPCA’s website lists artemether, arteether and artesunate under its products and also lists artemether and artesunate as having EDMF and WHO certificates. The assignee also has Canada HPFB certificate for artemether.
The Central Drug Research Institute (CDRI) in collaboration with IPCA is developing CDRI-97/78 (1,2,4 trioxane derivative), a synthetic artemisinin substitute for treating drug resistant Plasmodium falciparum infection. In July 2016, CDRI-97/78 was reported to be in phase 1 clinical development. IPCA in collaboration with CDRI was also investigating CDRI-99/411, a synthetic artemisinin substitute for treating malaria; but its development had been presumed to have been discontinued; however, this application’s publication would suggest otherwise.
Writeup
Artemisinin is an active phytoconstituent of Chinese medicinal herb Artemisia annua, useful for the treatment of malaria. Generally, artemisinin/artemisinic acid is obtained by extraction of the plant, Artemisia annua. The plant Artemisia annua was first mentioned in an ancient Chinese medicine book written on silk in the West Han Dynasty at around 200 B.C. The plant’s anti-malarial application was first described in a Chinese pharmacopeia, titled “Chinese Handbook of Prescriptions for Emergency Treatments,” written at around 340 A.D.
Artemisinin being poorly bioavailable limits its effectiveness. Therefore semisynthetic derivatives of artemisinin such as artesunate, dihydroartemisinin, artelinate, artemether, arteether have been developed to improve the bioavailability of Artemisinin.
Artemisinin and its derivatives – dihydroartemisinin, artemether, arteether, and artesunate being a class of antimalarials compounds used for the treatment of uncomplicated, severe complicated/cerebral and multi drug resistant malaria. Additionally, there are research findings that artemisinin and its derivatives show anti-parasite, anti-cancer, and anti-viral activities.
Dihydroartemisinin Artesunate
The content of Artemisinin in the plant Artemisia annua varies significantly according to the climate and region/geographical area where it is cultivated. Further, the extraction methods provide artemisinin or artemisinic acid from the plant in very poor yields and therefore not sufficient to accommodate the ever-growing need for this important drug. Consequently, widespread use of these valuable drugs has been hampered due to the low availability of this natural product. Therefore, research has focused on the syntheses of this valuable drug in a larger scale to meet the increasing global demand and accordingly ample literature is available on the synthesis of artemisinin or its derivatives, but no commercial success being reported / known till date.
Artemisinin can be prepared synthetically from its precursors such as artemisinic acid or dihydroartemisinic acid according to literature methods known to skilled artisans. For example, dihydroartemisinic acid can be converted to artemisinin by a combination of photooxidation and air-oxidation processes as described in U.S. Patent No. 4,992,561.
Amorphadiene is an early starting material for synthesis of Artemisinic acid or dihydroartemisinic acid, which is an important intermediate for producing Artemisinin commercially, and WO2006128126 reported a preparation method as mentioned in scheme- 1.
acid
In accordance with the scheme 1, the amorphadiene is treated with di(cyclohexyl)borane ( δΗι ΒΗ followed by reaction with H2O2 in presence of NaOH to obtain the amorph-4-ene 12-ol which is further oxidized to dihydroartemisinic acid using CrCb/ifcSC^. The formation of amorph-4-ene 12-ol is taking place via epoxidation of the exocyclic double bond. However, the reported yields of this synthesis are very low, making it unviable to produce artemisinic acid at a cheaper cost than natural extraction, for commercial use.
Amorpha -4, 11-diene
A similar method is published in, WO2009088404, for synthesis of dihydroartemisinic acid through preparation of amorph-4-ene-12-ol via epoxide formation, albeit, predominantly at exo position by reacting the amorpha-4,11-diene with H2O2 in presence of porphyrin catalyst (TDCPPMnCl). During reaction, epoxidation also occurred at endo position leading to formation of Amorphadiene- 4,5- epoxide that remain as impurity. The formed exo epoxide (amorphadiene – 11, 12 – epoxide) is further reduced to get amorph- 4-ene 12-ol and then converted to dihydroartemisinic acid and finally converted into artemisinin.
Amorphadiene-11,12-epoxide
This process involves expensive & industry unfriendly reagents. Moreover, desired stereo isomers were obtained only in poor yields, because several purification steps were needed to get desired stereo isomers leading to escalated production/operational costs.
Therefore there remains a need in the art to improve the yield of Dihydroartemisinic acid, which could potentially reduce the cost of production of Artemisinin and/or its derivatives. Consequently it is the need of the hour to provide a synthetic and economically viable process to meet the growing worldwide demand by improving the process for Artemisinin and/or its derivatives to obtain them in substantially higher yields with good purity by plant friendly operations like crystallization/extractions rather than column chromatography/other cost constraint procedures.
Therefore, the object of the invention is to prepare Artemisinic acid of formula-II, Dihydroartemisinic acid of formula-IIa, Artemisinin and its derivatives through Amorphadiene- 4,5- epoxide.
DHAA methyl ester
Scheme 2
Method 4 (From compound of formula IV (R = CI)):
In the 4-neck round bottom flask was charged Diphenyl sulfoxide (23.8 g), NaHC03 (32.96 g) and DMSO (80 ml) at 30°C. Further a solution of compound of formula IV (R = CI) (10 g) in DMSO (20 ml) was charged to the reaction mass at 30°C followed by heating and maintaining the temperature for 40 hours at 80°C till completion. DMSO was distilled out under vacuum. The reaction mass was cooled followed by charging water
(100 ml) and toluene (100 ml) to the reaction mass with stirring for 30 minutes at 28°C. The layers were separated out and aqueous layer was back extracted with toluene (2 X 100 ml). The organic layer was washed with water (100 ml) and saturated brine solution (100 ml). Solvent was distilled out under vacuum at 50°C, and the crude mass degassed under vacuum at 50-55°C. IPA (40 ml) was charged to the mass. Simultaneous addition of hydrazine hydrate (65% in aqueous solution) (3.8 g) and hydrogen peroxide (50% in aqueous solution) (2.5 ml) was done at 30-32°C over a period of 3.25 hours. After completion, reaction mass was cooled up to 5-10°C and water (100ml) was added to the reaction mass. The pH of the reaction mass was adjusted to 3.8 with dilute 8% aqueous HC1 (24 ml) at 10°C. Ethyl acetate (60 ml) was added to the reaction mass at 10°C and stirred for 15 minutes at 15-20°C. The layers were separated. Aqueous layer was back extracted with ethyl acetate (2 X 20 ml). The combined organic layer was washed with 10%) sodium metabisulfite solution (50 ml), water (50 ml) and saturated brine solution (50 ml). The organic layer was distilled out under vacuum at 45°C and the obtained crude mass was degassed at 50-55°C. To this was added DME (40 ml), Biphenyl (0.9 g) and Li-metal (1.63 g) and the reaction mass was maintained for 10 hours at 80-85°C till reaction completion. The reaction mass was cooled up to 0-5°C followed by drop wise addition of water within one hour, and the reaction stirred for two hours at 20-25°C. Toluene (35 ml) was charged with stirring and layers were separated. The aqueous layer was washed with toluene (35 ml) and the combined toluene layer was washed with water (20 ml). The combined aqueous layer was again washed with toluene (20 ml). The aqueous layer was cooled to 10-15°C and pH adjusted to 3.5-4 with dilute 16% aqueous HC1. MDC (50 ml) was charged and stirred 30 minutes at 20-25°C followed by separation of layers. The aqueous layer extracted with MDC (25 ml) and the combined MDC layer was washed with water (50 ml), then with saturated NaCl solution (25 ml). The solvent was distilled out under vacuum at 40-45°C and the crude mass (Purity: 70-80%>) was degassed at 65-70°C. The crude product (10 g) was dissolved in ethyl acetate (200 ml). 10%> aqueous NaOH (100 ml) was charged to the reaction mass and stirred one hour at 20°C followed by layer separation. Again 10%> aqueous NaOH (100ml) was added to the organic layer, stirred for 30 minutes and layers were separated out. The pH of the combined NaOH solution wash was adjusted to 4.0 with dilute 16%> aqueous HC1 at 5-10°C under stirring. Ethyl acetate (850 ml) was charged to aqueous acidic mass, stirred 30 minutes and layers were separated out. The aqueous layer was back extracted with ethyl acetate (2 X 30 ml) and the combined organic layer was washed with water (100 ml) and saturated brine (50 ml). The organic layer was dried over sodium chloride, solvent was distilled out under vacuum and the purified mass was degassed under vacuum at 50-55°C to obtain Dihydroartemisinic acid (Purity: 90-95%).
b) Methyl ester of Dihydroartemisinic acid:
To a clear solution of Dihydroartemisinic acid (40 g) dissolved in MDC (120 ml) was added thionyl chloride (SOCh) (14.85 ml) at 10±2°C and reaction mass was heated to reflux temperature 40±2°C. After the completion of reaction, solvent was distilled out and excess SOCh was removed under reduced pressure. The resulting concentrated mass of acid chloride was dissolved in MDC (200 ml). In another RBF was taken triethylamine (30.6 ml) and methanol (120 ml). To this solution was added above acid chloride solution at 30±2°C and maintained till completion of reaction. To the reaction mass was added water (400 ml) and organic layer was separated. The aqueous layer was washed with MDC and mixed with main organic layer and the combined organic layer was back washed with water till neutral pH. Then organic layer was concentrated to give methyl ester of Dihydroartemisinic acid as a brown color oily mass.
Weight: 41.88 gm
Yield = 98%
c) Artemisinin:
Methyl ester of dihydroartemisinic acid (67.7 g) was dissolved in methanol (338 ml). To this solution was added Sodium molybdate (29.5 g), 50% hydrogen peroxide (147.3 g) was added at 30±2°C and reaction was maintained for 3-4 hours. After completion of reaction was added water (300 ml) and MDC (300 ml) to the reaction mass. The organic layer was separated and aqueous layer washed with MDC (100 ml). The combined organic layer was concentrated to 475 ml containing hydroperoxide intermediate and directly used for next stage reaction. In another RBF containing MDC (475 ml) was added benzene sulfonic acid (1.27 g) and Indion resin (6.7 g). This heterogeneous solution was saturated with oxygen by passing O2 gas for 10 min at 0±2°C. To this was added previous stage hydroperoxide solution at same temperature with continuous 02 gas purging within 30-40 minutes. The oxygen gas was passed at same temp for 4 hours and temperature raised to 15±2°C with continued passing of oxygen for 5 hours. The
mixture was stirred at 25-30°C for 8-10 hours followed by filtration of resin. The filtrate was washed with water (200 ml X 3) and the combined aqueous layer back washed with MDC (50 ml). The combined organic layer was concentrated to give crude Artemisinin. Weight: 54 gm
Yield= 70.7%
Purification of Artemisinin:
Crude Artemisinin (10 g) was dissolved in ethyl acetate (25 ml) at 45-50°C. The solution was cooled to 30-35°C followed by addition of n-Hexane (100 ml). The material was isolated, stirred for 2 hours, filtered and vacuum dried at 45°C.
Weight: 4 gm
Yield: 40%
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, [email protected], +91 9323115463 India
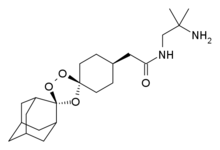
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2′ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4′-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
UncategorizedComments Off on Sanofi and PATH launch large-scale malaria drug production
Aug202014
Sanofi and global health charity PATH have come together to launch a large-scale production line of malaria jab semisynthetic artemisinin at Sanofi’s Garessio site in Italy.
Global demand for artemisinin, the key ingredient of artemisinin-based combination therapies (ACTs) for malaria, has increased since the World Health Organization identified ACTs as the most effective malaria treatment available.
Because the existing botanical supply of artemisinin – derived from the sweet wormwood plant – is inconsistent, having multiple sources of high-quality product will strengthen its supply chain, contribute to a more stable price, and ultimately ensure greater availability of treatment to people suffering from malaria, according to Sanofi.
INDIAComments Off on Ranbaxy obtains approval to market malaria drug in India
Oct232013
Ranbaxy Laboratories has secured approval from India’s Central Drugs Standard Control Organisation (CDSCO) to produce and market its Synriam drug in the country to treat malaria caused by the Plasmodium vivax parasite in adults.










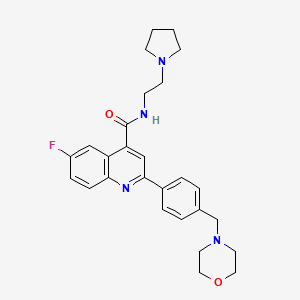 DDD498
DDD498