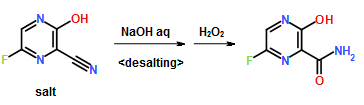Vorinostat
Zolinza, SAHA, suberoylanilide hydroxamic acid, Suberanilohydroxamic acid, N-hydroxy-N’-phenyloctanediamide
US patent 5369108, PDT PATENT
For the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma who have progressive, persistent or recurrent disease on or following two systemic therapies. Inhibits histone deacetylase I & 3.
- CCRIS 8456
- HSDB 7930
- M344
- N-Hydroxy-N’-phenyloctanediamide
- SAHA
- SAHA cpd
- Suberanilohydroxamic acid
- suberoylanilide hydroxamic acid
- UNII-58IFB293JI
| N-hydroxy-N‘-phenyl-octanediamide | |
|---|---|
| Trade names | Zolinza, 100 MG, CAPSULE, ORAL |
| ZOLINZA (VORINOSTAT) [Merck Sharp & Dohme Corp.] | |
| MedlinePlus | a607050 |
| Licence data | US FDA:link |
| LAUNCHED 2006 MERCKhttp://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf | |
| Legal status | ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Protein binding | 71% |
| Metabolism | Hepatic glucuronidation andoxidation CYP system not involved |
| Half-life | 2 hours |
| Excretion | Renal (negligible) |
| Identifiers | |
| CAS number | 149647-78-9 |
| ATC code | L01XX38 |
| Chemical data | |
| Formula | C14H20N2O3 |
| Mol. mass | 264.32 g/mol |
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=Vorinostat
Vorinostat (rINN) also known as suberanilohydroxamic acid (suberoyl+anilide+hydroxamic acid abbreviated as SAHA) is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities.
Vorinostat is marketed under the name Zolinza for the treatment of cutaneous T cell lymphoma (CTCL) when the disease persists, gets worse, or comes back during or after treatment with other medicines.[1] The compound was developed by Columbia University chemist, Ronald Breslow.
Vorinostat was the first histone deacetylase inhibitor[2] approved by the U.S. Food and Drug Administration (FDA) for the treatment of CTCL on October 6, 2006. It is manufactured by Patheon, Inc., in Mississauga, Ontario, Canada, for Merck & Co., Inc., White House Station, New Jersey.[3]
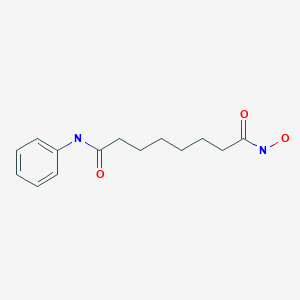
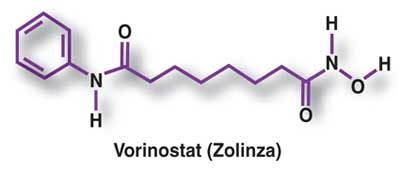
ZOLINZA contains vorinostat, which is described chemically as N-hydroxy-N’-phenyloctanediamide. The empirical formula is C14H20N2O3. The molecular weight is 264.32 and the structural formula is:
 |
Vorinostat is a white to light orange powder. It is very slightly soluble in water, slightly soluble in ethanol, isopropanol and acetone, freely soluble in dimethyl sulfoxide and insoluble in methylene chloride. It has no chiral centers and is non-hygroscopic. The differential scanning calorimetry ranged from 161.7 (endotherm) to 163.9°C. The pH of saturated water solutions of vorinostat drug substance was 6.6. The pKa of vorinostat was determined to be 9.2.
Each 100 mg ZOLINZA capsule for oral administration contains 100 mg vorinostat and the following inactive ingredients: microcrystalline cellulose, sodium croscarmellose and magnesium stearate. The capsule shell excipients are titanium dioxide, gelatin and sodium lauryl sulfate.
Vorinostat has been shown to bind to the active site of histone deacetylases and act as a chelator for Zinc ions also found in the active site of histone deacetylases [4] Vorinostat’s inhibition of histone deacetylases results in the accumulation of acetylated histones and acetylated proteins, including transcription factors crucial for the expression of genes needed to induce cell differentiation. [4]
SAHA inhibits class I and class II HDACs at nanomolar concentrations and arrests cell growth in a wide variety of transformed cells in culture at 2.5-5.0 µM. This compound efficiently suppressed MES-SA cell growth at a low dosage (3 µM) already after 24 hours treatment. Decrease of cell survival was even more pronounced after prolonged treatment and reached 9% and 2% after 48 and 72 hours of treatment, respectively. Colony forming capability of MES-SA cells treated with 3 µM vorinostat for 24 and 48 hours was significantly diminished and blocked after 72 hours.
Vorinostat has also been used to treat Sézary syndrome, another type of lymphoma closely related to CTCL.[5]
A recent study suggested that vorinostat also possesses some activity against recurrent glioblastoma multiforme, resulting in a median overall survival of 5.7 months (compared to 4 – 4.4 months in earlier studies).[6] Further brain tumor trials are planned in which vorinostat will be combined with other drugs.
Including vorinostat in treatment of advanced non-small-cell lung cancer (NSCLC) showed improved response rates and increased median progression free survival and overall survival (although the survival improvements were not significant at the P=0.05 level).[7]
It has given encouraging results in a phase II trial for myelodysplastic syndromes in combination with Idarubicin and Cytarabine.[8]
Vorinostat is an interesting target for scientists interested in eradicating HIV from infected persons.[9] Vorinostat was recently shown to have both in vitro and in vivo effects against latently HIV infected T-cells.[10][11]
Vorinostat, represented by structural formula (I) and chemically named as N-hydroxy-N’- phenyl-octanediamide or suberoylanilide hydroxamic acid (SAElA), is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities and vorinostat is marketed, under the brand name Zolinza®, for the treatment of a type of skin cancer called cutaneous T-cell lymphoma (CTCL). Vorinostat is approved to be used when the disease persists, gets worse, or comes back during or after treatment with other medicines. Vorinostat has also been used to treat Sέzary’s disease and, in addition, possesses some activity against recurrent glioblastoma multiforme.

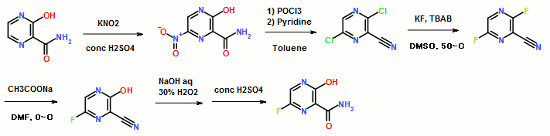
Vorinostat was first described in US patent 5369108, wherein four different synthetic routes for the preparation of vorinostat are disclosed (Schemes 1 to 4).
The single step process illustrated in Scheme 1 involves coupling of the diacid chloride of suberic acid with aniline and hydiOxylamine hydrochloride. However, the yield of this reaction is only 15-30%.

Scheme 1
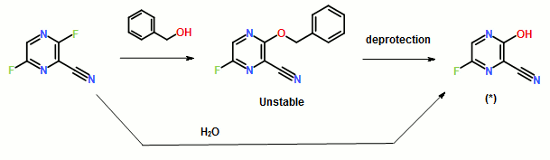
The multistep process illustrated in Scheme 2 begins with the monomethyl ester of suberic acid, which undergoes conversion to the corresponding acid chloride. Further coupling with aniline gives the methyl ester of suberanilic acid. Hydrolysis of the ester and further coupling with benzyl protected hydroxylamine gives benzyl protected vorinostat which on deprotection gives vorinostat.
HO. (CH2J6 OMe . ,OOMM e
O O



Scheme 2
In addition to the disadvantage of being a five-step process with overall yields reported as 35-65%, this process suffers from further disadvantages such as the use of the expensive monomethyl ester of suberic acid.

Scheme 3
The two step process illustrated in Scheme 3 involves coupling of the diacid chloride of suberic acid with aniline and O-benzyl hydroxylamine and then deprotection. However, the overall yield of this reaction is only 20-35%.

Scheme 4
The process illustrated in Scheme 4 is similar to that illustrated in Scheme 3, with the exception that O-trimethylsilyl hydroxylamine was used instead of O-benzyl hydroxylamine. The overall yield of this reaction is reported as 20-33%.
Another process for the preparation of vorinostat has been reported in J. Med. Chem.,
1995, vol. 38(8), pages 1411-1413. The reported process, illustrated in Scheme 5, begins with the conversion of suberic acid to suberanilic acid by a high temperature melt reaction.
Suberanilic acid is further converted to the corresponding methyl ester using Dowex resin and the methyl ester of suberanilic acid thus formed is converted to vorinostat by treatment with hydroxylamine hydrochloride. However, this process employs high temperatures (1900C) in the preparation of vorinostat which adds to the inefficiency and high processing costs on commercial scale. The high temperatures also increase the likelihood of impurities being formed during manufacture and safety concerns. The overall yield reported was a poor 35%.

MeOH, Dowex, 22 hours


Scheme 5
Another process for the preparation of vorinostat has been reported in OPPI Briefs, 2001, vol. 33(4), pages 391-394. The reported process, illustrated in Scheme 6, involves conversion of suberic acid to suberic anhydride, which on treatment with aniline gives suberanilic acid. Coupling of this suberanilic acid with ethyl chloroformate gives a mixed anhydride which upon treatment with hydroxylamine gives vorinostat in an overall yield of 58%. In the first step, there is competition between the formation of suberic anhydride and the linear anhydride and consequently isolation of pure suberic anhydride from the reaction mixture is very difficult. This process step is also hindered by the formation of process impurities and competitive reactions. In the second step, there is formation of dianilide by reaction of two moles of aniline with the linear anhydride. In the third step, suberanilic acid is an inconvenient by-product as the suberanilic acid is converted to a mixed anhydride with ethyl chloroformate, which is highly unstable and is converted back into suberanilic acid. Consequently, it is very difficult to obtain pure vorinostat from the reaction mixture. Although the reported yield was claimed to be 58%, when repeated a yield of only 38% was obtained.

Scheme 6
A further process for the preparation of vorinostat has been reported in J. Med. Chem., 2005, vol. 48(15), pages 5047-5051. The reported process, illustrated in Scheme 7, involves conversion of monomethyl suberate to monomethyl suberanilic acid, followed by coupling with hydroxylamine hydrochloride to afford vorinostat in an overall yield of 79%. However, the process uses the expensive monomethyl ester of suberic acid as starting material.
HOBt, DCC, DMF, RT, 4 hours



…………………….
VORINOSTAT
http://www.google.com/patents/EP2349985A2
A preferred embodiment of the first aspect of the present invention is illustrated in Scheme

suberic acid subefanilic acid NH2OHHCl, CDI

suberoylanilide hydroxamic acid (T)
Scheme 8
Optionally, an activating agent can be used in step (a) and/ or step (b) to afford products with high yields and purity. Preferably, the activating agent is selected from cyanuric chloride, cyanuric fluoride, catecholborane, or a mixture thereof. The activating agent is preferably used in combination with the coupling agent. A preferred embodiment of the process according to the first aspect of the present invention comprises the following steps:
(i) taking a mixture of THF, CDI and DCC;
(ii) adding suberic acid; (iii) adding aniline in THF to the solution from step (ii);
(iv) stirring at 25-30°C;
(v) filtering off the solid dicyclohexyl urea formed in the reaction;
(vi) concentrating the filtrate in vacuo;
(vii) adding a solution of KOH in water; (vϋi) filtering off the solid by-product;
(ix) heating the filtrate;
(x) adding aq. HCl;
(xi) isolating suberanilic acid;
(xii) mixing the suberanilic acid and CDI in DMF; (xiii) adding hydroxylamine hydrochloride as solid to the mixture from step (xii);
(xiv) isolating vorinostat from the mixture obtained in step (xiii);
(xv) adding acetonitrile and aq. ammonia to the vorinostat from step (xiv);
(xvi) heating the mixture;
(xvii) cooling the mixture to 20-27°C; and (xvϋi) isolating pure vorinostat from the mixture obtained in step (xvii).
Preferably, by utilising the same organic solvent in steps (a) and (b), pure vorinostat can be obtained without isolation of any synthetic intermediate^).
A preferred embodiment of the second aspect of the present invention is illustrated in Scheme 9.

suberic acid N-hydtoxy-7-carboxy-heptanamide

Example 1
Stage 1 : Conversion of suberic acid to suberanilic acid
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 3O0C. Suberic acid (leq) and aniline (leq) in THF (1 vol) was added and the mixture stirred for a further 16-20 hours. The solid by-product was removed by filtration and the filtrate was concentrated in vacuo at 5O0C. The solid residue obtained was treated with a solution of KOH (2eq) in water (10 vol) and stirred for 30 minutes at 25-300C and any solid byproduct formed was removed by filtration. The filtrate obtained was heated at 6O0C for 3-4 hours and cooled to 200C before addition of an aqueous solution of HCl (17.5%, 3 vol). The mixture was stirred for 30 minutes and the solid filtered, washed with water (2×5 vol) and dried under vacuum at 60-650C. Molar Yield = 60-65% Purity by HPLC = 99.5%
Stage 2: Conversion of suberanilic acid to crude vorinostat The suberanilic acid (leq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2eq) was added at 25-3O0C and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2×5 vol) and dried under vacuum at 500C. Molar Yield = 70-75% Purity by HPLC = 99% Stage 3: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-30°C. The mixture was then maintained at 55-60°C for 1 hour before being cooled to 20-25°C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-5O0C for 5 hours. Molar Yield = 55-60% Purity by HPLC > 99.8%
Example 2
Stage 1 : Conversion of suberic acid to crude vorinostat
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 30°C. Suberic acid (leq) and hydroxylamine (leq) in THF (1 vol) was added and the mixture stirred for a further 1 hour. Then CDI (0.5eq), DCC (0.8eq) and aniline (leq) were added to the mixture and the mixture was stirred for a further 16-20 hours. The solid byproduct was removed by filtration and the filtrate was concentrated in vacuo at 50°C to obtain crude vorinostat. Molar Yield = 55-60% Purity by HPLC > 95.8%
Stage 2: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-3O0C. The mixture was then maintained at 55-600C for 1 hour before being cooled to 20-250C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-500C for 5 hours. Molar Yield = 35-40% Purity by HPLC > 99.8%
…………………………………….
SYNTHESIS
Scheme V. – –

Vorinostat
Suberic acid (l.Oeq) was dissolved in tetrahydrofuran (15vol) and the clear solution was chilled to 0-5°C. Methyl chloro formate (l.leq) and triethylamine (1.1 eq) were added to the solution at the same temperature and the mixture was stirred for 15 minutes. The triethylamine.HCl salt formed was filtered off, then aniline (leq) was added to the reaction mixture at 0-50C and stirring was continued for 15 minutes. Methyl chloroformate (l.leq) and triethylamine (l.leq) were added to the clear solution and stirring was continued for a further 15 minutes at 0-5°C. This chilled reaction mixture was added to a freshly prepared hydroxylamine solution in methanol (*see below) chilled to 0-5°C and stirred for 15 minutes at 0-5°C. The solvent was removed under vacuum at 40°C and the residue obtained was taken in methylene dichloride and the organic solution was washed with water and dried over anhydrous sodium sulfate. Methylene dichloride was removed under vacuum at 40°C and acetonitrile was added to the residue. This mixture was stirred for 15 minutes before the solid was filtered under vacuum and dried under vacuum at 60°C to afford the product as a white solid. Molar yield = 35-41%; HPLC purity = 99.90%.
VORINOSTAT
1H-NMR (DMSO-d6): 1.27 (m, 4H, 2 x -CH2-), 1.53 (m, 4H, 2 x -CH2-), 1.94 (t, J = 7.3 Hz, 2H, -CH2-), 2.29 (t, J = 7.4 Hz, 2H, -CH2-), 7.03 (t, J = 7.35 Hz, IH, aromatic para position), 7.27 (t, J = 7.90 Hz, 2H, aromatic meta position), 7.58 (t, J = 7.65 Hz, 2H, aromatic ortho position), 8.66 (s, IH, -OH, D2O exchangeable), 9.85 (s, IH, amide -NH-, D2O exchangeable), 10.33 (s, IH, -NH-OH, D2O exchangeable).
13C-NMR (DMSO-d6): 25.04 (2C, 2 x -CH2-), 28.43 (2C, 2 x -CH2-), 32.24 (1C, -CH2-), 36.34 (1C, -CH2-), 119.01 (2C, Ar-C), 122.96 (1C, Ar-C), 128.68 (2C, Ar-C), 139.24 (1C, Ar- C, =CNH-), 169.23 (1C, -CO-), 171.50 (1C, -CO-).
*Preparation of hydroxylamine solution:
Potassium hydroxide (l.leq) was added to methanol (8vol) and the solution was chilled to 0-5°C. Similarly hydroxylamine hydrochloride (l.leq) was added to methanol (8vol) and chilled to 0-5°C. The chilled amine solution was added to the chilled alkali solution and stirred for 15 minutes at 0-50C. The white potassium chloride salt was filtered off and the filtrate was used as such.
SPECTRAL DATA AND SYNTHESIS
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for structures see above link
Suberoylanilide hydroxamic acid (26, SAHA, vorinostat).
Suberic acid monomethyl ester (23) (15.09 g, 80.2 mmol) and DMF (0.10 mL) in anhydrous
DCM (300 mL) was added SOCl2 (34.6 mL, 0.481 mol), and the reaction mixture was refluxed for 3
h. The mixture was then concentrated. Toluene (300 mL) was added to the residue and evaporated
to afford crude acid chloride 24. Crude 24 was dissolved in DCM (240 mL), and followed by
addition of aniline (7.3 mL, 80.2 mmol) and Et3N (16.9 mL, 0.120 mol). The reaction mixture was
stirred for 90 min at room temp. The course of reaction was monitored by TLC (30% EtOAc in
hexanes) and LC–MS. DCM was removed, and ethyl acetate (500 mL) was added to dissolve the
residue. The organic layer was washed with aqueous NaHCO3 (500 mL × 2), 1 N HCl (400 mL × 2),
water, dried (Na2SO4), and evaporated to dryness under reduced pressure. The residue was purified
by vacuum liquid chromatography (silica, 20% EtOAc in hexanes) to afford compound 25as white crystalline solids (20.15 g, 96 %). NaOMe in MeOH solution (5.4 M, 106 mL, 0.573 mol) was added to a solution of compound 25 (10.05 g, 38.2 mmol) and NH2OH·HCl (26.54 g, 0.382 mol) in
dry MeOH (375 mL). The reaction mixture was stirred for 40 min at room temp. The reaction was
quenched by adding of 1 N HCl to pH 7–8. MeOH was removed under reduced pressure and water
(1 L) was added to the residue. The precipitated solid was filtered and washed with water (300 mL)
and EtOAc (150 mL) to afford crude 26 which was further purified by recrystallization. MeOH (200
mL) was added to crude 26 (5 g) and warmed to dissolve all solids. The MeOH solution was filtered,
and deionized water (400 mL) was added to the filtrate, the resulting solution was placed at 4 oC
overnight. Crystals obtained were filtered and washed with deionized water (100 mL) to afford pure
26 (vorinostat, SAHA) as off-white crystals. Overall yield: 80–85% from compound 23. Compound
26,
LC–MS m/z 265.1 ([M + H]+).
1H NMR (DMSO-d6) 10.35 (1H, s), 9.86 (1H, s), 8.68 (1H, s),
7.58 (2H, d, J = 7.6 Hz), 7.28 (2H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.4 Hz), 2.29 (2H, t, J = 7.4 Hz),
1.94 (2H, t, J = 7.4 Hz), 1.57 (2H, m), 1.49 (2H, m), 1.33 – 1.20 (2H, m); 13C NMR (DMSO-d6)
171.2, 169.1, 139.3, 128.6, 122.9, 119.0, 36.3, 32.2, 28.4, 28.3, 25.0. Anal. (C10H20N2O3) C, H, N.
References
- “ZOLINZA, Merck’s Investigational Medicine for Advanced Cutaneous T-Cell Lymphoma (CTCL), To Receive Priority Review from U.S. Food and Drug Administration” (Press release). Merck & Co. June 7, 2006. Retrieved 2006-10-06.
- HDAC Inhibitors Base (vorinostat)
- “FDA Approves New Drug for Skin Cancer, Zolinza” (Press release). Food and Drug Administration. October 6, 2006. Retrieved 2006-10-06.
- Richon, Victoria. “Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor”. British Journal of Cancer. Retrieved 3 May 2012.
- Cuneo A, Castoldi. “Mycosis fungoides/Sezary’s syndrome”. Retrieved 2008-02-15.
- “Vorinostat shows anti-cancer activity in recurrent gliomas” (Press release). Mayo Clinic. June 3, 2007. Retrieved 2007-06-03.
- http://www.rtmagazine.com/reuters_article.asp?id=20091209clin013.html Dec 2009. URL dead Jan 2012
- “Zolinza, Idarubicin, Cytarabine Combination Yields High Response Rates In MDS Patients (ASH 2011)”.
- “Study of the Effect of Vorinostat on HIV RNA Expression in the Resting CD4+ T Cells of HIV+ Pts on Stable ART”. ClinicalTrials.gov. 2011-03-21.
- Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM (2009). “Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid.”. AIDS Res Hum Retroviruses 25 (2): 207–12. doi:10.1089/aid.2008.0191. PMC 2853863. PMID 19239360.
- Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J et al. (2009). “Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells.”. J Biol Chem 284 (11): 6782–9.doi:10.1074/jbc.M807898200. PMC 2652322. PMID 19136668.
- Vorinostat bound to proteins in the PDB
- J. Med. Chem.,1995, vol. 38(8), pages 1411-1413.
- A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells
J Med Chem 2005, 48(15): 5047 - A new facile and expeditious synthesis of N-hydroxy-N’-phenyloctanediamide, a potent inducer of terminal cytodifferentiation
Org Prep Proced Int 2001, 33(4): 391 - US patent 5369108, PDT PATENT
- WO2007/22408………
- WO 1993007148
- CN 102344392
| United States | 7456219 | APPROVAL 2006-11-14 | EXPIRY 2026-11-14 |
| United States | 6087367 | 1994-10-04 | 2011-10-04 |
| Canada | 2120619 | 2006-11-21 | 2012-10-05 |
| Patent | Patent Expiry | pat use code |
|---|---|---|
| 7399787 | Feb 9, 2025 | U-892 |
| 7456219 | Mar 11, 2027 | |
| 7652069 | Mar 4, 2023 | |
| 7732490 | Mar 4, 2023 | U-892 |
| 7851509 | Feb 21, 2024 | U-892 |
| 8067472 | Mar 4, 2023 | U-892 |
| 8093295 | May 16, 2026 | |
| 8101663 | Mar 4, 2023 | U-892 |
| RE38506 | Nov 29, 2013 |
U 892 =TREATMENT OF CUTANEOUS MANIFESTATIONS IN PATIENTS WTIH CUTANEOUS T-CELL LYMPHOMA (CTCL)
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ODE | Oct 6, 2013 |
| WO2009098515A1 * | Feb 6, 2009 | Aug 13, 2009 | Generics Uk Ltd | Novel process for the preparation of vorinostat |
Marks, P.A., Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotech 25(1) 84-90 (2007). DOI: 10.1038/nbt1272
Takashi Kumagai, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. International Journal of Cancer. 2007 Aug 1;121(3):656-65. DOI: 10.1002/ijc.22558
Hrzenjak A, et al. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer. 2010 Mar 4;9:49. DOI: 10.1186/1476-4598-9-49
………………………………………………………………………………………
| US7148257 | Aug 26, 2003 | Dec 12, 2006 | Merck Hdac Research, Llc | Methods of treating mesothelioma with suberoylanilide hydroxamic acid |
| US7375137 | Mar 28, 2006 | May 20, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7399787 | Jul 9, 2003 | Jul 15, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7456219 | Jun 19, 2003 | Nov 25, 2008 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7652069 | Oct 30, 2007 | Jan 26, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7732490 | Sep 11, 2007 | Jun 8, 2010 | Merck Hdac Research, Llc | Methods of treating cancer |
| US7847122 | Mar 18, 2008 | Dec 7, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7851509 | Mar 18, 2008 | Dec 14, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7879865 | Nov 18, 2005 | Feb 1, 2011 | Sloan-Kettering Institute For Cancer Research | Treatment of cancer of the brain using histone deacetylase inhibitors |
| US7998957 | Feb 6, 2008 | Aug 16, 2011 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicylcoheptenes, their preparation and use |
| US8058268 | Jul 29, 2009 | Nov 15, 2011 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8067472 | Apr 23, 2010 | Nov 29, 2011 | Merck Hdac Research, Llc | Methods of treating Hodgkin’s and non-Hodgkin’s lymphoma |
| US8088951 | Nov 30, 2007 | Jan 3, 2012 | Massachusetts Institute Of Technology | Epigenetic mechanisms re-establish access to long-term memory after neuronal loss |
| US8093295 | May 16, 2006 | Jan 10, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing the same |
| US8101663 | Dec 7, 2009 | Jan 24, 2012 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US8143445 | Oct 1, 2008 | Mar 27, 2012 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8227473 | Jul 17, 2009 | Jul 24, 2012 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8288440 * | Jan 13, 2010 | Oct 16, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8329719 | Aug 1, 2011 | Dec 11, 2012 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8426444 | Jun 30, 2011 | Apr 23, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8450372 * | Jan 13, 2010 | May 28, 2013 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8455688 | Mar 21, 2012 | Jun 4, 2013 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8541458 | Jun 11, 2012 | Sep 24, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8563615 | Nov 1, 2010 | Oct 22, 2013 | Massachusetts Institute Of Technology | Use of CI-994 and dinaline for the treatment of memory/cognition and anxiety disorders |
| US20100112046 * | Jan 13, 2010 | May 6, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100113829 * | Jan 13, 2010 | May 6, 2010 | Cote Aaron S | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100119596 * | Jan 13, 2010 | May 13, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20110263712 * | Oct 14, 2009 | Oct 27, 2011 | Generics (Uk) Limited | Process for the preparation of vorinostat |
| US20110313044 * | Jun 16, 2011 | Dec 22, 2011 | Urquima S.A. | Polymorphs of Suberoylanilide Hydroxamic Acid |
| EP2079304A1 * | Sep 24, 2007 | Jul 22, 2009 | Merck & Co., Inc. | Amine base salts of saha and polymorphs thereof |
| EP2229941A1 * | May 16, 2006 | Sep 22, 2010 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| EP2292221A2 * | May 16, 2006 | Mar 9, 2011 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127319A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127321A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2008039421A2 * | Sep 24, 2007 | Apr 3, 2008 | Arlene E Mckeown | Pharmaceutical compositions of hdac inhibitors and chelatable metal compounds, and metal-hdac inhibitor chelate complexes |
| WO2008042146A1 * | Sep 24, 2007 | Apr 10, 2008 | Arlene E Mckeown | Amine base salts of saha and polymorphs thereof |
| WO2008097654A1 * | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| WO2009020565A1 * | Aug 1, 2008 | Feb 12, 2009 | Lixte Biotechnology Inc | Use of phosphatases to treat neuroblastomas and medulloblastomas |
| WO2010061220A2 * | Nov 25, 2009 | Jun 3, 2010 | Generics [Uk] Limited | Novel processes and pure polymorphs |
EXTRAS
MS-275 (Entinostat); CI-994 (Tacedinaline); BML-210; M344; MGCD0103 (Mocetinostat); PXD101 (Belinostat); LBH-589 (Panobinostat); Tubastatin A; Scriptaid; NSC 3852; NCH 51; HNHA; BML-281; CBHA; Salermide; Pimelic Diphenylamide; ITF2357 (Givinostat); PCI-24781; APHA Compound 8; Droxinostat; SB939.



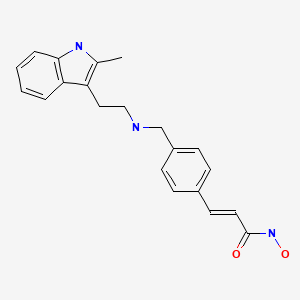 panobinostat
panobinostat













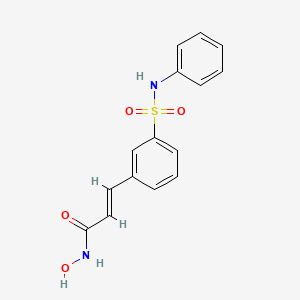 BELINOSTAT
BELINOSTAT







































 CLAZOSENTAN
CLAZOSENTAN CLAZOSENTAN DI-NA SALT is discontinued
CLAZOSENTAN DI-NA SALT is discontinued





















 rapamycin
rapamycin SIROLIMUS
SIROLIMUS











 SIROLIMUS
SIROLIMUS



 MIDOSTAURIN
MIDOSTAURIN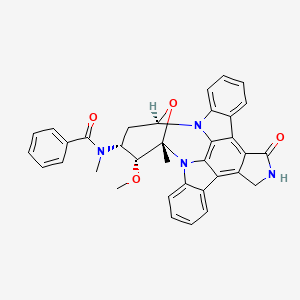 MIDOSTAURIN
MIDOSTAURIN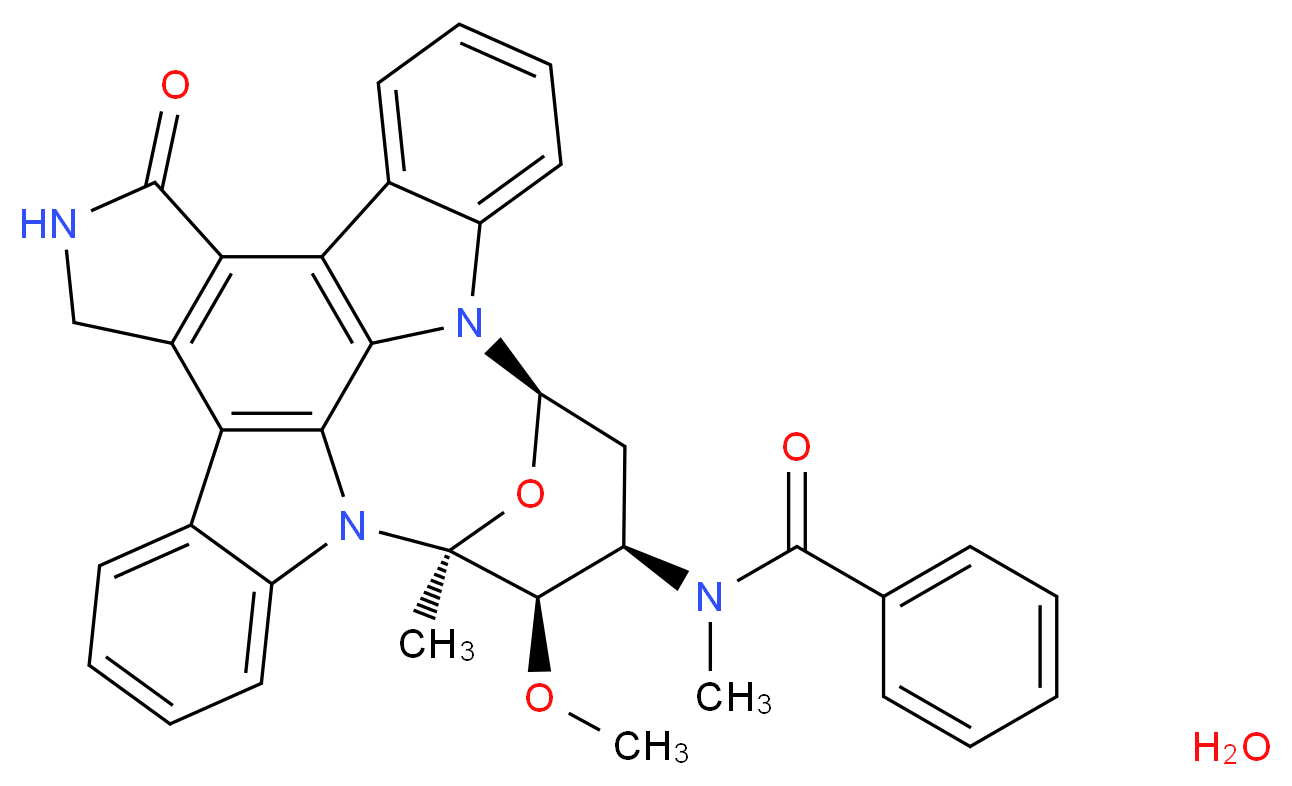






 cicaprost
cicaprost cicaprost
cicaprost