Investigational New Drug Application













This table provides links to information for investigators about submitting Investigational New Drug (IND) applications to FDA. The resources for application reporting and applications procedures apply to IND applications for both clinical research and clinical treatment.
| IND Applications for Clinical Investigations(Product Development) |
IND Application Reporting |
IND Application Procedures |
IND Applications for Clinical Treatment(Expanded Access) |
|---|---|---|---|
| Overview | Overview | Overview | Overview |
| Contents and Format | Protocol Amendments |
Exemptions from IND Requirements | Contents and Format |
| Regulatory and Administrative Components |
Information Amendments |
Interactions with FDA |
Emergency IND Timeline (Treatment of a Single Patient in Emergency Setting) |
| Non-clinical Components |
Safety Reports |
Clinical Hold | For Physicians: A Guide to Non-emergency Single Patient Expanded Access Submissions |
| Clinical Components |
Annual Reports |
Investigator’s Responsibilities |
Treatment of a Group of Patients |
The United States Food and Drug Administration‘s Investigational New Drug (IND) program is the means by which a pharmaceutical company obtains permission to start human clinical trials and to ship an experimental drug across state lines (usually to clinical investigators) before a marketing application for the drug has been approved. Regulations are primarily at 21 CFR 312. Similar procedures are followed in the European Union, Japan, and Canada
Introduction
An investigational new drug is a new drug or biological drug that is used in a clinical investigation. This term also includes biological products used in vitro for diagnostic purposes.The Investigational New Drug Application (IND) is a request for an exemption from the federal statute that prohibits an unapproved drug from being shipped in interstate commerce.
IND is not an application for marketing authorization of a drug. For the purpose of this lesson, “IND” is synonymous with “Notice of Claimed Investigational Exemption for a New Drug.”Upon completion of preclinical study and collection of data, the sponsor (the person who takes responsibility for and initiates a clinical investigation) must submit an application to FDA to notify FDA it is conducting a clinical study on human subjects.
This application is called an Investigational New Drug Application (INDA or IND).Investigational new drug application abbreviated as INDA is a mandatory requirement filed with the FDA in order to seek permission for administering a new drug under investigation to Human subjects after completion of the preclinical studies on it.
2. It confers protection to the subjects and also sees that the investigational plan is efficient and designed to achieve its required objectives.
3. The sponsor of the drug is responsible for the initiation of clinical trials.
4. He might be an individual i.e. sponsor-investigator, a pharmaceutical company, governmental agency, academic institution or private or public organization.
5. The INDA is filed with the FDA under Title 21, Code of Federal Regulations Section 312- the guidelines for preapproval of all clinical testing’s are specified.
| Regulation of therapeutic goods in the United States |
|---|
 |
| Prescription drugs Over-the-counter drugs |
6. CONTENT OF INDA
[1] (i). All the requirements for submitting an INDA are prescribed in the Code of Federal Regulations and are submitted under a cover sheet (Form FDA- 1571) (1).
(ii). The items required are: – Name, address and telephone number of the sponsor of the drug. – Name and title of the person responsible for monitoring the conduct and progress of the investigation. – Names and titles of the persons responsible for the review and evaluation of information relevant to the safety of the drug. – Name and address of any contract research organization involved in the study (if any). –
Identification of the phase/phases of clinical investigation to be conducted. – Introductory statement and general investigational plan including, name of drug and all active ingredients, structural formula and pharmacological class, formulation, route of administration, and broad objectives and planned duration of study. – Description of the investigational plan.
The reason for selecting a drug or research study. Indications to be studied, approach to evaluation of the drug, types of studies, estimated number of subjects to be given the drug, and any risks anticipated based on animal studies. – Brief summary of previous experience with the drug, including reasons if the drug is withdrawn. –
Chemistry and manufacturing control information like physical, chemical and biologic characteristics, product stability during the clinical investigation. – Pharmacological and Toxicological information. –
If the drug is a combination of previously investigated components, then preclinical and clinical data of these components when given singly and in combination. – Clinical protocol for planned study. –
Commitment that an “Institutional Review Board” (IRB) has approved the study and will continue to monitor the investigation. – Investigators brochure. – Commitment not to begin clinical investigations until IND is in effect signature of the sponsor or authorized representative and the date of signed application.
7. After submission of IND to the FDA, it is assigned to the various divisions of Center for Drug Research and Evaluation wing of the FDA for its review and evaluation.
8. The FDA has 30days from the receipt of IND to decide whether the proposed clinical trial should proceed or not. If the sponsor is not contacted within 30days, the trial may proceed.
9. Meanwhile reviewers at FDA may put a “Clinical Hold” on the proposed Clinical trials at any time. This prevents human testing of drug. It may be due to the following reasons:[2] – If there is any unreasoned threat to the safety of the trial subjects i.e. if the subjects face any illness or injury due to treatment. – Insufficient data to assess patient risks. – If the investigators involved in the study do not meet the necessary requirements with respect their qualification. – Misleading or incomplete investigators brochure.
10. In case if all the requirements for FDA approval are satisfied an IND is granted. Once an IND is in effect, all the proposed changes to the original IND thereafter, must be submitted as amendments for approval.
References:
1. Ansel’s Pharmaceutical Dosage forms and Drug delivery systems, 8th Edition, Pg: 44-45.
2. The Science and Practice of Pharmacy: Remington Pharmaceutical Sciences, 21st Edition, Volume 1, Pg: 965-967, Published By Lippincott William & Wilkins. “This Blog Contains No Plagiarized Material”
more info
IND Format and Content
.: Section 1. Application Form and Coversheet
Form 1571
Form 1571 can be obtained from the FDA Web site.
Please go to the » FDA Web site help page for instructions on how to fill out form 1571.
Coversheet
The coversheet must have basic information about applicant (name and address), submission contents, and must be signed and dated by the sponsor.
» Click here to see the Coversheet template.
Section 2. Table of ContentsEach application must also have a table of contents, with page and volume number, to make it easy to find information.
» Click here to see the Table of Contents template.
Section 3. Introductory Statement The introductory statement is a brief, two- or three-page summary of the entire IND. Its intent is to help FDA understand drug development plans and sponsor needs.
The statement should include:
- The name of the drug;
- Structure ;
- Pharmacological class;
- Development history;
- Information regarding active ingredient(s) ;
- Dosage form;
- Route of administration;
- Planned duration of the study;
- Foreign testing;
- Previous human experience in other countries that may be relevant; and
- Any withdrawal from the market.
Section 4. General Investigational Plan
The general investigation plan briefly describes the development plan for the following year.
The plan should include:
- The rationale for the drug or the study;
- Indications to be studied;
- A summary of clinical studies to be conducted in the first year;
- The estimated number of subjects; and
- Anticipated risk based on toxicological data or prior human studies.
Section 5. Investigator’s Brochure
The investigator’s brochure should include:
- Brief description of drug and formulation including the structural formula;
- Summary of pharmacological and toxicological effects in animals;
- Summary of pharmaco-kinetics and biological disposition in animals and (if known) in humans;
- Summary of safety and effectiveness in humans (if any); and
- Possible risks and side effects
Early brochures will have more preclinical data; later versions will contain more clinical data.
Section 6. Protocol
Initially, a protocol for each planned study should be submitted in the original IND. In general, protocol for Phase I studies may be less detailed and more flexible than protocols for Phase II and III.
The protocol for Phase I should outline:
- The investigation;
- Number of patients to be involved;
- Description of the safety exclusions; and
- Description of dosing plan (including duration, dose, or methods to be used in determining dose).
Section 7. Chemistry, Manufacturing, and Control
This section should provide information regarding thecomposition, manufacture, and control of the drug substance and the drug product for different phases of the study. The amount of information included will depend on the scope of the study.
Provide detailed information for each of the following:
- Drug substance;
- Drug product;
- Placebo;
- Labeling; and
- Environmental impact.
Section 8. Pharmacology and Toxicology
This section should provide adequate information
about pharmacological and toxicological studies
of the drug involving laboratory animals or in vitro, on
the basis of which the sponsor has concluded that it is
reasonably safe to conduct the proposed clinical investigations.
This section of the submission shall include:
- Information regarding identification/qualification of the individuals who evaluated the non-clinical data;
- Statement of where investigations were conducted and where records will be available for inspection;
- Summaries of preclinical studies of pharmacological and toxicological effects and mechanism(s) of action:
- Pharmacological effects and mechanism of action related to proposed therapeutic use in animals;
- Other pharmacological effects relevant to safety; and
- Any information available on Absorption, Distribution, Metabolism, and Excretion (ADME) and rationale for dose selection also should be submitted in this section.
Integrated summary of toxicological effects in animals and in vitro studies. It should include the results of acute, subacute, and chronic toxicity tests; tests of the drug’s effects on reproduction and the developing fetus; any special toxicity test related to the drug’s particular mode of administration or conditions of use (e.g., inhalation, dermal, or ocular toxicology); and any in vitro studies intended to evaluate drug toxicity. A tabulation of data suitable for detailed review should be submitted to support the safety of the drug.
The extent of the information depends on the class of drug and the intended phase of the clinical study.
Toxicology reports can be submitted in a draft format if the final report is not ready at the time of submission. The final report can be submitted when submitting the IND annual report.
Non-clinical studies should be conducted in compliance with GLP regulations. If not, a statement should be made explaining the reason for the noncompliance.
» Click here to see the Animal and Clinical Study Protocol template.
Section 9. Previous Human Experience
This section should contain a summary of known previous human experience in the United States and other countries that support the investigational drug
Also include:
- Data from other INDs and NDAs;
- Marketing information or withdrawal from any market; and
- Published information on safety or efficacy.
Section 10. Additional Information
This section contains information on special topics, such as drug dependence and abuse potential, radioactive drugs, pediatric studies, and plans for assessing pediatric safety and effectiveness.
Any other information that will help the evaluation of the clinical investigations with respect to their safety or to their design and potential as controlled clinical trials to support marketing of the drug should be included in this section.
Section 11. Relevant Information
Any other information relevant to the study needed for review of the application should be incorporated in this section.
This may include:
- Minutes of FDA meetings (if there has been any Pre-IND meeting);
- Steps taken for the FDA suggestion;
- Reference to previously submitted information;
- Literature and publications in foreign language and their English translations; and
- Any other relevant information.
IND Review Process
When the FDA receives the original IND, it assigns an IND number. This is the number that the applicant or sponsor should use in all correspondence with FDA regarding the application.
The application is then forwarded to the appropriate reviewing division. The reviewing division will send a letter to the sponsor-investigator providing notification of the IND number assigned, date of receipt of the original application, the address where future submissions to the IND should be sent, and the name and telephone number of the FDA representative to whom questions about the application should be directed.
INDs are not approved, they become effective 30 days after receipt by FDA
if clinical hold (suspension or delay in clinical investigations) is not imposed.
Unless notified earlier by the FDA that studies may begin, the sponsor should
wait for 30 days to initiate clinical studies.
clinical Hold
A clinical hold is an order from the FDA to suspend or delay a proposed clinical investigation or to suspend an ongoing investigation. The most common reasons for a clinical hold include:
- Unreasonable risk to subjects;
- Investigator not qualified;
- Investigator brochure misleading;
- Insufficient information to assess risk;
- Gender bias in a study in patients with life-threatening disease; and
- Deficient study design
IND Maintenance
Once the IND is in effect, the sponsor must amend it as needed
to ensure that current information is submitted to FDA and
that FDA is aware of the changes. The three main sections we
will look at in this topic are Amendments (protocol and information),
IND Safety Reports, and Annual Reports.
Protocol Amendments
There are three types of protocol amendments:
- New Protocol: For studies that are not covered by protocol already contained in the IND.
- Change in protocol: To explain changes to the current protocol that significantly affect the safety of the subjects, the scope of the investigation, and scientific quality of the study (e.g., increase in drug dose, addition or dropping of a control group).
- New investigator: When a new investigator is added to carry out a previously submitted protocol. The sponsor should notify FDA of the new investigator within 30 days of investigator being added.
The content and format of a protocol amendment:Clearly identify the amendment (e.g., “Protocol Amendment: New protocol” or Protocol Amendment: Change in Protocol”);
- Form 1571;
- In the case of new protocol, a copy of the new protocol and a brief description of significant difference from the previous protocol;
- In the case of new investigator, name and the qualifications (CV) and reference to the previous protocol; and
- Reference, if necessary, to specific technical information in the IND or submitted information.
Information Amendment
Report any change that is not within the scope of protocol amendment, IND safety report, or annual reports under the information amendment. Examples include new toxicology, chemistry, other technical information, or discontinuation of a clinical investigation.
An information amendment can be submitted as necessary, but not more frequently than every 30 days.
The content and format of the information amendment:
Clearly identify the amendment with the information submitted (e.g., if it is CMC information, it shall bear the title, “Information Amendment: “Chemistry, Manufacturing and Control” or “Information Amendment: Pharmacology-Toxicology”);
- Form 1571;
- An statement explaining the nature and the purpose of the submission;
- The organized records and reports; and
- Organized data (if necessary).
IND Safety Reports |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Annual ReportsThe annual report is sent to FDA to update the IND on the investigation progress and all changes that are not reported in amendments or other reports. Annual reports should be submitted within 60 days of the date that the IND went into effect.
and
|
||||||||||||||||||||||||||||||||||||||||||
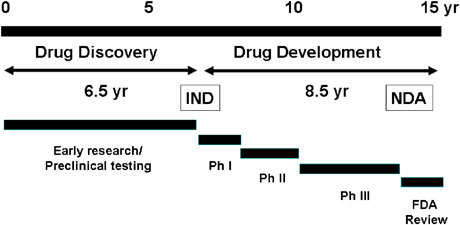
Types

Timeline for drug evaluation
- Commercial INDs are filed by companies to obtain marketing approval for a new drug.
- Research or investigator INDs are non-commercial INDs filed by researchers to study an unapproved drug or to study an approved drug for a new indication or in a new patient population.
- Emergency Use INDs, also called compassionate use or single-patient INDs, are filed for emergency use of an unapproved drug when the clinical situation does not allow sufficient time to submit an IND in accordance with 21 CFR §§ 312.23, 312.24. These are most commonly used for life-threatening conditions for which there is no standard treatment.
- Treatment INDs are filed to make a drug available for treatment of serious or immediately life-threatening conditions prior to FDA approval. Serious diseases or conditions are stroke, schizophrenia, rheumatoid arthritis, osteoarthritis, chronic depression, seizures, Alzheimer’s dementia, amyotrophic lateral sclerosis (ALS), and narcolepsy.
- Screening INDs are filed for multiple, closely related compounds in order to screen for the preferred compounds or formulations. The preferred compound can then be developed under a separate IND. Used for screening different salts, esters and other drug derivatives that are chemically different, but pharmacodynamically similar.
Application
The IND application may be divided into the following categories:[1]
- Preclinical testing consists of animal pharmacology and toxicology studies to assess whether the drug is safe for testing in humans. Also included are any previous experience with the drug in humans (often foreign use).
- Manufacturing Information includes composition, manufacturer, and stability of, and the controls used for, manufacturing the drug. Used to ensure that the company can adequately produce and supply consistent batches of the drug.
- Investigator information on the qualifications of clinical investigators, that is, the professionals (generally physicians) who oversee the administration of the experimental drug to the study subjects. Used to assess whether the investigators are qualified to fulfill their clinical trial duties.
- Clinical trial protocols are the centerpiece of the IND. Detailed protocols for proposed clinical studies to assess whether the initial-phase trials will expose the subjects to unnecessary risks.
- Other commitments are commitments to obtain informed consent from the research subjects, to obtain review of the study by an institutional review board (IRB), and to adhere to the investigational new drug regulations.
An IND must also include an Investigator’s Brochure intended to educate the trial investigators of the significant facts about the trial drug they need to know to conduct their clinical trial with the least hazard to the subjects or patients.
Once an IND is submitted, the FDA has 30 days to object to the IND or it automatically becomes effective and clinical trials may begin. If the FDA detects a problem, it may place a clinical hold on the IND, prohibiting the start of the clinical studies until the problem is resolved.
Experimental drugs under an IND must be labeled “Caution: New Drug – Limited by Federal (or United States) law to investigational use.”
Prevalence
Approximately two-thirds of both INDs and new drug applications (NDAs) are small-molecule drugs. The rest is biopharmaceuticals. About half of the INDs fail in preclinical and clinical phases of drug development.
Examples
The FDA runs a medical marijuana IND program (the Compassionate Investigational New Drug program). It stopped accepting new patients in 1992 after public health authorities concluded there was no scientific value to it, and due to President George H.W. Bush administration’s desire to “get tough on crime and drugs.” As of 2011, four patients continue to receive cannabis from the government under the program.[2]
Sanctioned by Executive Order 13139, the US Department of Defense employed an anthrax vaccine classified as an investigational new drug (IND) in its Anthrax Vaccine Immunization Program (AVIP).

References
- ^ John S. McInnes (2011), “New Drug Applications”, in Shayne C. Gad (ed.), Pharmaceutical Sciences Encyclopedia, doi:10.1002/9780470571224.pse420
- ^ “4 Americans get medical pot from the feds”. CBS News. September 28, 2011.
External links
- Investigational New Drug (IND) Application Process Center for Drug Evaluation and Research, Food and Drug Administration.
- ICH Guidance for Industry, E6 Good Clinical Practice: Consolidated Guidance. BROKEN LINK
- Troetel, W.M.: Achieving a Successful US IND Filing (1) The Regulatory Affairs Journal. 6: 22–28, January 1995.
- Troetel, W.M.: Achieving a Successful US IND Filing (2) The Regulatory Affairs Journal. 6: 104–108, February 1995.
- Henninger, Daniel (2002). “Drug Lag”. In David R. Henderson (ed.). Concise Encyclopedia of Economics (1st ed.). Library of Economics and Liberty. OCLC 317650570, 50016270, 163149563
- IND Forms and Instructions from the US Food and Drug Administration
///////////
33,554 total views, 2 views today
