Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
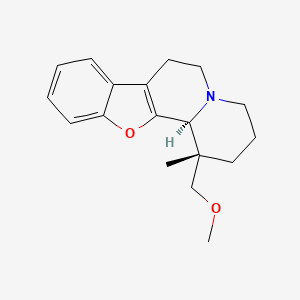
The basic drug substance candidate ORM10921 (MW = 285.38),
IUPAC name [1R*,12bR*)-(−)-1,3,4,6,7,12b-hexahydro-1-methoxymethyl-1-methyl-2H-benzofuro [2,3-a]quinolizine],
and its hydrochloric salt were synthesized by Orion Pharma, Finland.
The absolute configuration was assigned by optical rotation and later by single-crystal X-ray diffraction (see Supporting Information). The optical purity of the material was >97%.
Originator Juvantia Pharma (CEASED); Orion
Class Neuropsychotherapeutics
Mechanism of Action Alpha 2c adrenergic receptor antagonists
Highest Development Phases
Discontinued Major depressive disorder; Schizophrenia
Most Recent Events
10 May 2006 Discontinued – Phase-I for Schizophrenia in Finland (unspecified route)
10 May 2006 Discontinued – Preclinical for Depression in Finland (unspecified route)
15 Nov 2002 Preclinical trials in Schizophrenia in Finland (unspecified route)
Figure 1: Chemical structure of the study compound. Molecular Formula: C18H23NO2 · HCl · ½ H2O; Molecular Weights: 285.39 (free base), 321.85 (hydrochloride) 330.86 (hydrochloride hemihydrate). ORM-10921 · HCl is a single stereoisomer with the (1R*,12bR*) configuration.
The alpha adrenergic receptors can be divided on a pharmacological basis into alphal- and alpha2-adrenoceptors, which can both be further divided into subtypes. Three genetically encoded subtypes, namely alpha2A-, alpha2B- and alpha2C-adrenoceptors, have been discovered in human. Accordingly, alpha2- adrenoceptors in humans have been subdivided into three pharmacological subtypes known as alpha2A-, alpha2B- and alpha2C-adrenoceptors. A fourth, pharmacologically defined subtype, alpha2D, is known in rodents and in some other mammals, and it corresponds to the genetically defined alpha2A-adrenoceptors.
The alpha2-adrenoceptor subtypes have distinct tissue distributions and functional roles. For instance, while alpha2A-adrenoceptors are widely expressed in various tissues, alpha2C-adrenoceptors are concentrated in the CNS, and they appear to play a role in the modulation of specific CNS-mediated behavioural and physiological responses. Compounds that are non-specific to any of the above-mentioned alpha2 subtypes, and compounds that are specific to certain alpha2 subtypes, are already known. For example, atipamezole is a non-specific alpha2 antagonist. Atipamezole has been described in, for example, EP-A-183 492 (cf. p.13, compound XV) and Haapalinna, A. et al., Naunyn-Schmiedeberg’s Arch. Pharmacol. 356 (1997) 570-582. U.S. Patent No. 5,902,807 describes compounds that are selective antagonists for the alpha2C subtype and may be used in the treatment of mental illness, e.g. mental disturbance induced by stress. Such compounds include, for example, MK-912 and BAM- 1303. Furthermore, WO-A-99 28300 discloses substituted imidazole derivatives having agonist-like activity for alpha2B- or 2B/2C-adrenoceptors. hi addition, WO 01/64645 relates to derivatives of quinoline useful as alpha2 antagonists, as well as to selective alpha2C antagonist agents. The disclosures of all documents cited above in this paragraph are incorporated by reference herein.
Several arylquinolizine derivatives and related compounds have been described in the literature, some of which possess valuable pharmaceutical effects. For example, U.S. Patents No. 4,806,545 and 4,044,012 describe 1,1-disubstituted indolo[2,3-«]quinolizidines useful as vasodilators and antihypoxic agents. Further, substituted arylquinolizine derivatives, described for example in U.S. Patent No. 4,686,226 possessing alpha2-adrenoceptor antagonistic activity are useful for example as antidepressant, antihypertensive, or antidiabetic agents or platelet aggregation inhibitors. In addition, U.S. Patent No. 3,492,303 relates to indolo[2,3- α]quinolizidines useful as central nervous system depressants.
According to the European GMP-Rules, written procedures for tranfser activities and their documentation are required. For example, a Transfer SOP, a transfer plan and a report are now mandatory and will be checked during inspections.
As participant of the GMP education course “Product Transfer” in Berlin, from 25-27 October 2016 you will receive a special version of the Guideline Manager CD with a special section concerning product transfers. This section contains, amongst others, a Transfer SOP and a template for a Transfer Plan. Both documents are in Word format and can immediately be used after adoption to your own situation.
Regulatory Guidance Documents like the WHO guideline on transfer of technology in pharmaceutical manufacturing and the EU/US Variation Guidelines, are also part of the Guideline Manager CD. Due to copyright reasons, this CD is not available for purchase and can only be handed out to participants of the Product Transfer course.
regulatoryComments Off on Analytical Lifecycle: USP <1210> “Statistical Tools”, Analytical Target Profile and Analytical Control Strategy
Sep152016
Analytical Lifecycle: USP <1210> “Statistical Tools”, Analytical Target Profile and Analytical Control Strategy
The United States Pharmacopeia (USP) is currently undertaking further steps towards a comprehensive analytical lifecycle approach by publishing a draft of a new General Chapter <1210> Statistical Tools for Procedure Validation and two Stimuli Articles regarding Analytical Target Profile and AnalyticalControl Strategy in Pharmacopeial Forum. Read more about the life cycle concept for analytical procedures.
Following the recently announced elaboration of a new general chapter <1220> “The Analytical Procedure Lifecycle” the United States pharmacopeia (USP) is now proceeding in its approach for a comprehensive analytical lifecycle concept. A further step towards this approach is the draft of a new USP General Chapter <1210> Statistical Tools for Procedure Validation which has been published in Pharmacopeial Forum (PF) 42(5) in September 2016. Comment deadline is November 30, 2016.
Additionally, two Stimuli Articles regarding “Analytical Control Strategy” and “Analytical Target Profile: Structure and Application Throughout The Analytical Lifecycle” appeared in the same issue of the PF.
In the draft chapter <1210> Statistical Tools for Procedure Validation, the USP Statistics Expert Committee presents a revision to the proposal of <1210> published in PF 40(5) [Sept.–Oct. 2014]. On the basis of the comments and feedback given by stakeholders, the committee has addressed their concerns about the narrow scope and details on methodology to be used. The chapter is proposed as a companion to general chapter <1225> Validation of Compendial Procedures with the purpose of providing statistical methods that can be used in the validation of analytical procedures. A revision of general chapter <1225>, including a new section on Lifecycle Management of Analytical Procedures, has been published for comment in PF 42(2) in March 2016.
Specifically, the revision clarifies the accuracy and precision calculations while removing specific linearity requirements. Linearity may be inferred from accuracy or other statistical methods as deemed appropriate. The chapter discusses all of the following analytical performance characteristics from a statistical perspective:
accuracy,
precision,
range,
detection limit,
quantitation limit,
and linearity.
Additional related topics that are discussed in the draft include statistical power, two one-sided tests of statistical equivalence, tolerance intervals, and prediction intervals.
Furthermore, up to now, four Stimuli Articles regarding the analytical lifecycle have been published:
“Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification” in PF 39(5),
“Analytical Target Profile: Structure and Application Throughout The Analytical Lifecycle”.
The Analytical Target Profile (ATP) is the focal point of the lifecycle approach. It is comparable to the Quality Target Product Profile (QTPP) which is defined in ICH Q8. The Stimuli Article emphasizes “that the current approach to development, validation, verification, and transfer of analytical procedures has served the industry well.” The lifecycle approach – comprised of the development (design, stage 1), qualification (stage 2), and monitoring of the performance of analytical procedures (control strategy, stage 3) – is an extension of the current guidance, taking advantage of the learnings from ICH Q8 – quality by design (QbD) concepts. The Article considers in particular the following questions (and provides examples):
What is an ATP, and why is it useful?
How can the ATP criteria be established?
How can an ATP be applied during the three stages of the procedure lifecycle?
According to the article, “an additional advantage of using an ATP is that it can drive the development of a robust control strategy, resulting in better, more consistent performance of an analytical procedure throughout its lifecycle.”
In the Stimuli Article on the Analytical Control Strategy (ACS), the following questions are discussed:
What is the ACS?
What is the relationship between the ACS and the ATP?
What is the quality risk management (QRM) process and how can it be applied to an analytical procedure?
How does the ACS apply to the product lifecycle?
Additionally, examples of the following are provided:
How to develop an ACS using the QRM process, and
How to develop and apply a risk-based replicate strategy to minimize variability.
The USP Expert Panel would appreciate any feedback on the suggested approaches, as well as any alternative approaches for consideration.
Following your registration on the USP Pharmacopeial Forum website you can get to the proposal for general chapter <1210> and the complete stimuli articles. Because of the importance of the new USP chapter <1220>, ECA and USP join forces and organise the first joint event “Lifecycle Approach of Analytical Procedures“, in Prague, Czech Republic, from 8 to 9 November 2016.
The importance of Quality by Design (QbD) is being realized gradually, as it is gaining popularity among the generic companies. However, the major hurdle faced by these industries is the lack of common guidelines or format for performing a risk-based assessment of the manufacturing process. This article tries to highlight a possible sequential pathway for performing QbD with the help of a case study. The main focus of this article is on the usage of failure mode and effect analysis (FMEA) as a tool for risk assessment, which helps in the identification of critical process parameters (CPPs) and critical material attributes (CMAs) and later on becomes the unbiased input for the design of experiments (DoE). In this case study, the DoE was helpful in establishing a risk-based relationship between critical quality attributes (CQAs) and CMAs/CPPs. Finally, a control strategy was established for all of the CPPs and CMAs, which in turn gave rise to a robust process during commercialization. It is noteworthy that FMEA was used twice during theQbD: initially to identify the CPPs and CMAs and subsequently after DoE completion to ascertain whether the risk due to CPPs and CMAs had decreased.
Quality by Design in Action 1: Controlling Critical Quality Attributes of an Active Pharmaceutical Ingredient
Abstract: In this paper (R)-7-(azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride 1 was isolated and identified as the N-substituted regioisomer of besifloxacin, which has been synthesized from the reaction of 8-chloro-1-cyclopropyl-6,7-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 3 with (R)-tert-butyl 3-aminoazepane-1-carboxylate 2 in acetonitrile as solvent in 37% yield. The chemical structure of compound 1 was established on the basis of 1H-NMR, 13C-NMR, mass spectrometry data and elemental analysis.
Structural Characterization
1H-NMR (500 MHz, DMSO-d6): δ ppm: 14.73 (H-23, s, 1H), 9.72 (H-14, s, 2H), 8.69 (H-7, s, 1H),7.79 (H-1, d, J = 13.1 Hz, 1H), 6.20 (H-11, d, J = 9.1 Hz, 1H), 4.37 (H-12 and H-19, m, 2H), 3.38(H-13, m, 2H), 3.23 (H-15, m, 1H), 3.09 (H-15, m, 1H), 2.14 (H-18, m, 1H), 1.94 (H-16 and H-18, m,2H), 1.84 (H-16 and H-17, m, 2H), 1.60 (H-17, m, 1H), 1.23 (H-20 or H-21, m, 2H), 1.03 (H-20 orH-21, m, 2H).
13C-NMR(125 MHz, DMSO-d6): δ ppm: 175.6 (C-9), 165.4 (C-22), 151.7 (C-7), 150.6 (C-2), 148.7(C-3), 139.0 (C-5), 137.3 (C-4), 117.8 (C-10), 110.3 (C-1), 107.0 (C-8), 52.9 (C-12), 50.1 (C-13), 46.2(C-15), 41.3 (C-19), 34.0 (C-18), 24.9 (C-16), 21.6 (C-17), 10.9 (C-20 or C-21).
FAB-MS, m/z = 394.1 (M+).
Elemental analysis: Calculated for C19H21ClFN3O3.HCl: C, 53.03%; H, 5.15%; N, 9.77%; found: C,52.82%; H, 5.39%; N, 9.50%.
1H-NMR (500 MHz, DMSO-d6): δ ppm: 14.73 (H-23, s, 1H), 9.72 (H-14, s, 2H), 8.69 (H-7, s, 1H), 7.79 (H-1, d, J = 13.1 Hz, 1H), 6.20 (H-11, d, J = 9.1 Hz, 1H), 4.37 (H-12 and H-19, m, 2H), 3.38 (H-13, m, 2H), 3.23 (H-15, m, 1H), 3.09 (H-15, m, 1H), 2.14 (H-18, m, 1H), 1.94 (H-16 and H-18, m,2H), 1.84 (H-16 and H-17, m, 2H), 1.60 (H-17, m, 1H), 1.23 (H-20 or H-21, m, 2H), 1.03 (H-20 orH-21, m, 2H).
R&D Center, Jiangsu Yabang Pharmaceutical Group, Changzhou 213200, China
In this paper (R)-7-(azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride 1was isolated and identified as the N-substituted regioisomer of besifloxacin, which has been synthesized from the reaction of 8-chloro-1-cyclopropyl-6,7-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 3 with (R)-tert-butyl 3-aminoazepane-1-carboxylate 2in acetonitrile as solvent in 37% yield. The chemical structure of compound 1 was established on the basis of 1H-NMR, 13C-NMR, mass spectrometry data and elemental analysis
REGIOMER OF BESIFLOXACIN
BESIFLOXACIN
Zaixin Chen *
R&D Center, Jiangsu Yabang Pharmaceutical Group, Changzhou 213200, China
* Author to whom correspondence should be addressed;
Merck Serono, the biopharmaceutical business of Merck, and MMV announced today that an agreement has been signed for Merck Serono to obtain the rights to the investigational antimalarial compound DDD107498 from MMV. This agreement underscores the commitment of Merck Serono to provide antimalarials for the most vulnerable populations in need.
“This agreement strengthens our Global Health research program and our ongoing collaboration with Medicines for Malaria Venture,” said Luciano Rossetti, Executive Vice President, Global Head of Research & Development at Merck Serono. “MMV is known worldwide for its major contribution to delivering innovative antimalarial treatments to the most vulnerable populations suffering from this disease, and at Merck Serono we share this goal.”
DDD107498 originated from a collaboration between MMV and the University of Dundee Drug Discovery Unit, led by Prof. Ian Gilbert and Dr. Kevin Read. The objective of the clinical program is to demonstrate whether the investigational compound exerts activity on a number of malaria parasite lifecycle stages, and remains active in the body long enough to offer potential as a single-dose treatment against the most severe strains of malaria.
While development and commercialization of the compound is under Merck Serono’s responsibility, MMV will provide expertise in the field of malaria drug development, including its clinical and delivery expertise, and provide access to its public and private sector networks in malaria-endemic countries.
Merck Serono has a dedicated Global Health R&D group working to address key unmet medical needs related to neglected diseases, such as schistosomiasis and malaria, with a focus on pediatric populations in developing countries. Its approach is based on public-private partnerships and collaborations with leading global health institutions and organizations in both developed and developing countries.
“Working with partners like Merck Serono is critical to the progress of potential antimalarial compounds, like DDD107498, through the malaria drug pipeline,” said Dr. Timothy Wells, Chief Scientific Officer at MMV. “Their Global Health Program is gaining momentum and we need more compounds to tackle malaria, a disease that places a huge burden on the world’s most vulnerable populations. DDD107498 holds great promise and we look forward to working with the Merck Serono team through the development phase.”
According to the World Health Organization, there were an estimated 198 million cases of malaria worldwide in 2013, and an estimated 584,000 deaths, primarily in young children from the developing world. The launch of the not-for-profit research foundation, MMV, in 1999 and a number of collaborations and partnerships, including those with Merck Serono, has contributed to reducing the major gap in malaria R&D investment and subsequent dearth of new medicines.
“It’s hugely encouraging to see the German pharmaceutical industry increasing their engagement in the development of novel antimalarials,” said global malaria expert Prof. Dr. Peter Kremsner, Director of the Institute for Tropical Medicine at the University of Tübingen, Germany. “The Merck Serono and MMV collaboration to develop DDD107498 is a great step. It’s a compound that offers lots of promise so I’m excited to see how it progresses.
Scots scientists in ‘single dose’ malaria treatment breakthrough
STV
An antimalarial drug that could treat patients was discovered by Dundee university scientists
Scientists have discovered an antimalarial compound that could treat malaria patients in a single dose and help prevent the spread of the disease from infected people.
The compound DDD107498 also has the potential to treat patients with malaria parasites resistant to current medications, researchers say.
Scientists hope it could lead to treatments and protection against the disease, which claimed almost 600,000 lives amid 200 million reported cases in 2013.
The compound was identified through a collaboration between the University of Dundee’s drug discovery unit (DDU) and the Medicines for Malaria Venture (MMV), a separate organisation.
The compound is now undergoing further safety testing with a view to entering human clinical trials within the next year.
Details of the discovery have been published in the journal Nature.
Professor Ian Gilbert, head of chemistry at the DDU, who led the team that discovered the compound, said: “The publication describes the discovery and profiling of this exciting new compound.
“It reveals that DDD107498 has the potential to treat malaria with a single dose, prevent the spread of malaria from infected people and protect a person from developing the disease in the first place.
“There is still some way to go before the compound can be given to patients. However, we are very excited by the progress that we have made.”
The World Health Organisation reports that there were 200 million clinical cases of malaria in 2013, with 584,000 people dying from the disease. Most of these deaths were children under the age of five and pregnant women.
MMV chief executive officer Dr David Reddy said: “Malaria continues to threaten almost half of the world’s population – the half that can least afford it.
“DDD107498 is an exciting compound since it holds the promise to not only treat but also protect these vulnerable populations.
“The collaboration to identify and progress the compound, led by the drug discovery unit at the University of Dundee, drew on MMV’s network of scientists from Melbourne to San Diego.”The publication of the research is an important step and a clear testament to the power of partnership.”
MMV selected DDD107498 to enter preclinical development in October 2013 following the recommendation of its expert scientific advisory committee.
Since then, with MMV’s leadership, large quantities of the compound have been produced and it is undergoing further safety testing with a view to entering human clinical trials within the next year.
Merck Serono has recently obtained the right to develop and, if successful, commercialise the compound, with the input of MMV’s expertise in the field of malaria drug development and access and delivery in malaria-endemic countries.
Dr Michael Chew from the Wellcome Trust, which provides funding for the DDU and MMV, said: “The need for new antimalarial drugs is more urgent than ever before, with emerging strains of the parasite now showing resistance against the best available drugs.
“These strains are already present at the Myanmar-Indian border and it’s a race against time to stop resistance spreading to the most vulnerable populations in Africa.
“The discovery of this new antimalarial agent, which has shown remarkable potency against multiple stages of the malaria lifecycle, is an exciting prospect in the hunt for viable new treatments.”
PAPER
Discovery of a Quinoline-4-carboxamide Derivative with a Novel Mechanism of Action, Multistage Antimalarial Activity, and Potent in Vivo Efficacy
Conditions: (a) morpholine, Et3N, DCM, 16 h, 72% yield; (b) MeMgBr, toluene, reflux, 4 h and then a 10% aqueous HCl, reflux, 1 h, 70% yield; (c) NBS, benzoyl peroxide, dichlorobenzene, 140 °C, 16 h, 70% yield; (d) morpholine, K2CO3, acetonitrile, 40 °C, 16 h, 64% yield; (e) 5-fluoroisatin, KOH, EtOH, 120 °C, microwave, 20 min, 30–76% yield; (f) amine, CDMT, N-methylmorpholine, DCM, 20–61% yield.
A single-dose treatment against malaria worked in mice to cure them of the disease. The drug also worked to block infection in healthy mice and to stop transmission, according to a study published in Nature today. The fact that the drug can act against so many stages of malaria is pretty new, but what’s even more exciting is the compound’s mode of action: it kills malaria in a completely new way, researchers say. The feature would make it a welcome addition to our roster of antimalarials — a roster that’s threatened by drug resistance.
RESEARCHERS SIFTED THROUGH A LIBRARY OF ABOUT 4,700 COMPOUNDS TO FIND THIS ONE
Malaria is an infectious disease that’s transmitted through mosquito bites; it’s also a leading cause of death in a number of developing countries. Approximately 3.4 billion people live in areas where malaria poses a real threat. As a result, there were 207 million cases of malaria in 2012 — and 627,000 deaths. There are drugs that can be used to prevent malaria, and even treat it, but drug resistance is halting the use of certain treatments in some areas.
A long search
Searching for a new drug is all about trial and error. To find this particular compound, researchers sifted through a library of about 4,700 compounds, testing them to see if they were capable of killing the malaria parasite in a lab setting. When they found something that worked, they tweaked the drug candidate to see if it could perform more effectively. “We went through a lot of these cycles of testing and designing new compounds,” says Ian Gilbert, a medicinal chemist at the University of Dundee in the UK, and a co-author of the study. “Eventually we optimized to the compound which is the subject of the paper.” For now, that compound’s unwieldy name is DDD107498.
To make sure DDD107498 really had potential, the researchers tested it on mice that had already been infected with malaria. A single dose was enough to provoke a 90 percent reduction in the number of parasites in their blood. The scientists also gave the compound to healthy mice that were subsequently exposed to malaria. DDD107498 helped the mice evade infection with a single dose, but it’s unclear how long that effect would last in humans. Finally, the researchers looked at whether the compound could prevent the transmission from an infected mouse to a mosquito. A day after receiving the treatment, mice were put in contact with mosquitoes. The scientists noted a 91 percent reduction in infected mosquitoes.
“IT HAS THE ABILITY TO BE A ONE-DOSE [DRUG], IN COMBINATION WITH ANOTHER MOLECULE.”
“What’s exciting about this molecule is obviously the fact that it has the ability to be a one-dose [drug], in combination with another molecule to cure blood stage malaria,” says Kevin Read, a drug researcher also at the University of Dundee and a co-author of the study. The fact that the compound has the ability to block transmission and protect against infection is equally thrilling. But the way in which DDD107498 kills malaria might be its most interesting feature. It halts the production of proteins — which are necessary for the parasite’s survival. No other malaria drug does that right now, Read says. “So, in principle, there’s no resistance out there already to this mechanism.”
The drug hasn’t been tested in humans yet, so it may not be nearly as good in the field. But Read says DDD107498 looks promising. “From all the pre-clinical or non-clinical data we’ve generated, it is comparable or better than any of the current marketed anti-malarials in those studies.” And at $1 per treatment, the price of the drug should fall “within the range of what’s acceptable,” he says.
“It looks like an excellent study, and the results look very important,” says Philip Rosenthal, a malaria drug researcher at The University of California-San Francisco who didn’t participate in the study. This is a big shift for Rosenthal’s field. Five years ago, “we had very little going on in anti-malarial drug discovery,” he says. Now, there’s quite a bit going on for malaria researchers, and a number of promising compounds are moving along. DDD107498 “is another player, and it’s got a number of positive features,” he says.
OTHER TREATMENTS HAVE TO BE TAKEN FOR A FEW DAYS
One of the features is the drug’s potency. It’s very active against cultured malaria parasites, Rosenthal says. But what’s perhaps most intriguing about DDD107498 is that the drug works against the mechanism that enables protein synthesis the malaria parasite’s cells. No other malaria drug does that right now, Read says. “Considering challenges of treating malaria, which is often in rural areas and developing countries, a single dose would be a big plus,” he says. “In addition, because of it’s long half life, it may also work to prevent malaria with once a week dosing, which is also a benefit.”
Still, no drug is perfect. The data suggests that DDD107498 doesn’t kill malaria as quickly as some other drugs, Rosenthal says. And when the researchers tested it to see how long it might take for resistance to develop, the results weren’t as promising as he would like. The parasites figured out a way to become resistant to the compound “relatively easily,” he says. That shouldn’t be “deal-killer,” however. “Its slow onset of action probably means it should be combined with a faster-acting drug,” he says.
BUT IT’S SLOW-ACTING
The compound is going through safety testing now. If everything goes well, it should hit human trials within the next year, Read says. Chances are, it will have to be used in combination with other malaria drugs, Gilbert says. “All anti-malarials are given in combination because it slows down resistance.”
“When you’re treating infectious diseases, you know that drug resistance is always a potential problem, so having a number of choices to treat malaria is a good thing,” Rosenthal says. In this case, the drug’s new mode of action may hold lead to an entirely new weapon against malaria. “Obviously it’s got a long way to go,” Read says. But the compound is “very exciting,” nonetheless.
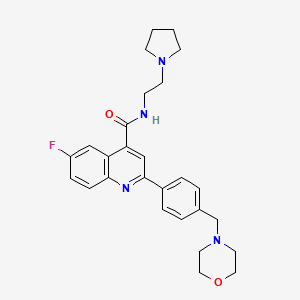
Example 16-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1 in Scheme 2
In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4-(morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130° C. under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHCO3 saturated aqueous solution (2×100 ml). The organic layer was separated, dried over MgSO4 and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10% B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60-200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50° C. for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
Example 26-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2
The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
Example 1AAlternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4
To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHCO3 saturated aqueous solution (2×100 ml) and the organic phase was separated, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10% B and then 15 min hold at 10% B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23% B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
Example 1 : 6-Fluoro-2-r4-(morpholinomethyl)phenyll-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide, Example compound 1 in Scheme 2
In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1- ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4- (morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130°C under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHC03 saturated aqueous solution (2 x 100 ml). The organic layer was separated, dried over MgS04and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10 % B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60- 200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50°C for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
Example 2: 6-Fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2
The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
Example 1A: Alternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2- pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4
To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro- 4,6-dimethoxy-1 ,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHC03 saturated aqueous solution (2x 100 ml) and the organic phase was separated, dried over MgS04 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10 %B and then 15 min hold at 10%B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23 % B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
A Quinoline Carboxamide Antimalarial Drug Candidate Uniquely Targets Plasmodia at Three Stages of the Parasite Life Cycle
Angewandte Chemie, International Edition (2015), 54, (46), 13504-13506
Putting a stop to malaria: Phenotypic screening against malaria parasites, hit identification, and efficient lead optimization have delivered the preclinical candidate antimalarial DDD107498. This molecule is distinctive in that it has potential for use as a single-dose cure for malaria and shows a unique broad spectrum of activity against the liver, blood, and mosquito stages of the parasite life cycle.
Prof. P. M. O’Neill Department of Chemistry, University of Liverpool Liverpool, L69 7ZD (UK) E-mail: [email protected] Prof. S. A. Ward Liverpool School of Tropical Medicine, Pembroke Place Liverpool, L3 5QA (UK)
Professor Ian Gilbert FRSC
Design and synthesis of potential therapeutic agents
Position:
Professor of Medicinal Chemistry and Head of the Division of Biological Chemistry and Drug Discovery
I am a medicinal chemist and my research interests are in the design and synthesis of potential drugs. The mainstay of my work is synthetic medicinal chemistry as part of the Drug Discovery Unit (DDU). Where possible we make extensive use of molecular modeling to guide our synthetic efforts. I have a particular interests in the following aspects of drug discovery:
Neglected diseases such as human African trypanosomiasis, leishmaniasis and malaria.
Chemical validation of drug targets, including novel targets for which there is little or no precedence for drug discovery.
Novel approaches to and paradigms for drug discovery.
Mode of action studies and target identification.
I am Head of Chemistry in the DDU. Our main focuses are on neglected diseases and novel drug targets. The neglected diseases we are tackling are malaria, tuberculosis and the kinetoplastid diseases. We use both target-based approaches and phenotypic approaches (whole parasite screening). We have had particular success in validating the enzyme N-myristoyltransferase as a drug target in human African trypanosomiasis, and in identifying and optimising phenotypic hits. In our novel targets area, we aim to validate novel areas of biology as potential drug targets.
spectroscopy, SYNTHESISComments Off on Application of On-Line NIR for Process Control during the Manufacture of Sitagliptin
Sep122016
The transamination-chemistry-based process for sitagliptin is a through-process, which challenges the crystallization of the active pharmaceutical ingredient (API) in a batch stream composed of multiple components. Risk-assessment-based design of experiment (DoE) studies of particle size distribution (PSD) and crystallization showed that the final API PSD strongly depends on the seeding-point temperature, which in turn relies on the solution composition.
To determine the solution composition, near-infrared (NIR) methods had been developed with partial least squares (PLS) regression on spectra of simulated process samples whose compositions were made by spiking each pure component, either sitagliptin free base (FB), water, isopropyl alcohol (IPA), dimethyl sulfoxide (DMSO), or isopropyl acetate (IPAc), into the process stream according to a DoE. An additional update to the PLS models was made by incorporating the matrix difference between simulated samples in lab and factory batches.
Overall, at temperatures of 20–35 °C, the NIR models provided a standard error of prediction (SEP) of less than 0.23 wt % for FB in 10.56–32.91 wt %, 0.22 wt % for DMSO in 3.77–19.18 wt %, 0.32 wt % for IPAc in 0.00–5.70 wt %, and 0.23 wt % for water in 11.20–28.58 wt %. After passing the performance qualification, these on-line NIR methods were successfully established and applied for the on-line analysis of production batches for compositions prior to the seeding point of sitagliptin crystallization.
Application of On-Line NIR for Process Control during the Manufacture of Sitagliptin
breakthrough designationComments Off on Shamisha Resource Managment, Experts in Recruitment, Pharmaceutical and FMCG Consulting
Sep092016
They specialise in Recruitment servicein Food/FMCG/ Pharma, with offices in Ahmedabad and Mumbai .We have track record of accurate and prompt service in most ethical manner. I happen to be a Start- up specialist too. I am myself PhD in Pharmaceutical Technology from IIT , BHU and have held top management positions in leadership role in P&G, Ranbaxy, Teva, Lupin, Novartis, JNJ,Colgate, Pfizer and can assure you of best recruitment solutions.
Shamisha Resources Management is founded by Technocrat Nina Sharma who holds PhD in Pharmaceutical Sciences from IIT , BHU and has worked in Corporate for 30 years as Technical Head including of Global R&D Centres of Topmost MNC,s.
Shamisha Resources Management is focused on Pharmaceutical Industry and has experise in recruitment service for India and global recruitment.
We have offices in Ahmedabad and Mumbai and have experienced and trained Recruitment consultants who work in most methodical and ethical manner. Our Service standards are very high in promptness and accuracy. We cover technical functions like R&D,QA, RA ,Manufacturing , Engineering, Project Management , IPR , Pharmacovigilance for both Formulations ,API and Sales and Marketing .We cover small molecules (ANDA) Speciality generics, biosimilars and large molecules. Our accuracy rate of fitment is 100%.Candidates are proposed only after thorough reference, background check and cultural fitment analysis as part of pre-screening programme at our end.
We are start-up specialists and support beyond recruitment solution in guiding on Organization structure and additional inputs for successful start up to operations.
M. Pharm ,PhD, Institute of Technology ,BHU
PGDMM , CIM UK
FULL RESUME
DR. NINA SHARMA
M.Pharm (Gold medalist)
PhD (Indian Institute of Technology, IIT, BHU, India), 1985
Post Graduate Diploma in Marketing Management (CIM, UK) 2007
Advanced Diploma in HR Management (ABE UK) 2008
Advanced Diploma in Business Management (ABE, UK) 2009
(Top Paper Prize Global Award for two papers)
FORMULATIONS EXPERT
Experienced in top level R&D Operations of Formulation Development, Global Portfolio and Resource management with successful projects completion and Formulation launches in various therapeutic categories, for OTC, ANDA and 505(b) (2), NCE and Biosimilars in reputed organizations with repeated track record of achievement
High level Formulation Development skill for LVP.SVP.Tablets, Capsules,Dry Syrup,Liquids, Semisolids for domestic and International markets, QA and Regulatory for USFDA , MCA and semi-regulated markets
Formulation development ,Technology transfer and Production operations ,Quality and compliance expert with track record of successful new products launches , portfolio expansion, Claims substantiation and Regulatory support, Tech transfer and supply chain transition management ,Project and team leadership to drive successful product launches within time and budget.
In recent roles as Senior Director with Johnson and Johnson ( J&J ) have led 10 million USD Formulation R &D project for Early developmentand Late Development ANDA ,manufacturing of clinical supplies for advanced market and Integrated drug product development for Emerging markets of China ,Korea, Taiwan and Mexico; Director Technical and Scientific Affairs with Teva Pharma Assignments with Novartis Healthcare Private Limited as R & D Head India – Global Research & Development Lab for OTC and Pfizer as Director (Head) for Formulation development Global R&D Center for Vet Medicine were successful projects in leadership position of Formulation R&D. Expertise in FMCG Sector as Head of Oral care Technology in India Global Formulation R&D Center, Procter and Gamble as R&D Manager Healthcare and Generics in Ranbaxy, Lupin and Searle.
Have published twenty research publications including four in international journals, US andEuropeanPatent holder across the illustrious career path.
Distinction of launching new products in conventional dosage forms, solids, liquids, sterile and semisolids and New Drug delivery systems covering full span of therapeutic area for infectious, psychiatry, cardiology, virology and biotechnology, pain and HIV, well versed with NDDS and Containment strategies.
An effective communicator with excellent people management/ training skills and strong analytical, problem solving & organizational abilities with proven track record of efficiently working with global partners.
CAREER HIGHLIGHTS
July 2015 to Present: Managing Director Shamisha Resources Management, Ahmedabad and Mumbai
Sept 2013 to July 2015– Director Technical Services and Production operation for Teva Pharma ( Procter &Gamble Teva Joint Venture PGT Healthcare ) , leading a team of 18 Managers /supervisors and Production staff of about 430 by end of 2014, handling commercialization of green field project of 600 crores.
Jan 2012 onwards Gresen Lehmann Group (GLG Singapore) consultant –successfully completed consultancy for Mckinsey, BCG on Pharmaceutical Development
July 2011 to Sep 2011 Senior VP Avesthagen Limited
July 2010 to July 2011 Senior Director Pharmaceutical Development and July 2008 to Feb 2010 Global Formulation R&D Center at Johnson and Johnson
The responsibilities included set up and start up of Pharmaceutical development organization for Chem Pharm , Global NCE Pre formulation and Late development for EU and US and support emerging markets of India , China , Brazil , Mexico .
Asia Drug product development strategy and capability calculations for three Sites, project allocation and transition plan, global SOP, s and processes.
Creating Road Map of Pharmaceutical Development till 2012 and integration
Since Sept 2005 Jan 2008 Novartis Healthcare Private Limited as Formulation R&D Head India – Global OTC Research &Development
Notable Deliverables
Led the effort in setting up the India Lab from Start up to operational build the team through recruitment and training
Head count increased to 47, established functions like Product development , Analytical, QA , Regulatory-CMC, Documentation ,CSV , I T Clinical operations , Facility Management , EHS ,Purchase and administration
The Stability centre having 18 chambers can cater to all the four zones including Brazil.
Use of LIMS, Empower, SAS and fully validated 21 CFR compliant systems.
Product design and submissions for ANDA in advanced market domain.
Clinical operations (Phase III) through CRO (Siro, Lotus, and Reliance Life Sciences) in the area of Generic OTC molecules for advanced market. Studies executed by Protocol preparation , approval ,quality audits and effective relation management , periodic reporting to global Clinical operations at Nyon ( Switzerland )
Played a key role in introducing Standard procedures (283 new) to make India Lab compliant with Global Quality operations standard and obtained USFDA Registration for the Site. The standards introduced by India Site are adopted by two other global sites. Data generation as per USFDA , EMEA , ANVISA and MCA
Efficiently led the Information Security and Safety Audits with successful outcome.
Distinction of leading the Group Quality Audit which is a tough internal standard to meet and India is the only compliant Lab so far.
Planning and control of Revenue and Capital budget for India Site
HOLDS THE MERIT OF
US FDA Registration ,Twenty eight Pharmaceutical development and Stability testing projects in the area Conventional Solids , Liquids semi solids and NDDS , Patches
Clinical operations, Technical writing, CMC Head count expansions within short time of lab operations.
Instrumental in collaborating with other Global Sites and local management to drive superior results.
Conducted successful studies on major global brands like Excedrin, Vibrocil, Theraflu, and Buckleys & Benefiber and applied the Project Management tools, innovative research based on market feedback. Milestones on delivery of Projects completed on time. Global project teams technical resource on global brands like Benefiber, Theraflu
Established a successful R&D Centre with high productivity and GMP within short time.
Apr 2004- August 20 05 Pfizer India Limited as Director
Planning and budgeting for new research center in Formulations and API , won 9 million dollar investment (first ever for the Thane Site )
Awarded four NCE Enhancement projects in Liquids and solids from Sandwich through bidding process for US and Europe market.
Formed the core team of R&D Scientists and Managers
Conducted early development studies for NCE molecules, coordinated pharmacokinetic studies on anthelmentics, Doramectin pour on, Selamectin with Praziquantel spot on formulations.
Technical liaison with Indian Regulatory Authorities for fast approvals of import of API and formulations.
Led the effort in establishing the Organization and formulating plans for expansion
Pivotal in establishing linkages across Pfizer, organization processes for reporting and communication, Project management and control systems, and people development.
Feb1999-March 2004 Colgate Palmolive India Ltd., General Manager-Oral Care
Notable Deliverables
Technical business support for driving growth in India, Pakistan, Africa and Middle East through new products launches in Oral care technology
Managed and coordinated largest Anti caries Clinical trials on Oral formulation with Triclosan and Fluoride conducted in India on 6000 children over a period of 24 months through Dentists
Managed and coordinated Multicentre Clinical trials on Whitening Oral care formulations conducted in India for Indian and Australian market
Managed and coordinated Clinical trials on formulations with different polishing agents
Managed and coordinated Clinical trials on Toothpowder and toothbrushes
Effective coordination with Global Research centre for Protocol preparation, approval, servicing of sample, reporting to Global
Efficiently liasioned with Government bodies for strategic partnership on future specifications for RM and Finished products pertaining to Oral care.
Led the effort to design, develop and Re launch major flagship brands Colgate Dental cream five times, Colgate toothpowder three times and Fresh Energy Gel two variants re-launched three times each to claim back the lost market shares (approximately 900 crores business). Regained the lost market share by nine point
Launching of Low cost formula with Cibaca brand name which resulted in increase in 7% Market share, formulation developed indigenously
Entry to medium price segment by launching Colgate Herbal
Led the efforts in the development of several innovative forms of Oral care including stripe formulations, innovative affordable oral care formulation.
Initiated several cost saving projects and implemented margin improvement programs on formulations.
Successfully obtained Ayurvedic classification to support pain claim for Herbal dentifrice due for launch.
Conducted several training programs for management team of manufacturing locations on Quality as certified trainer.
Spearheaded functions for entry into Low and medium price segments retaining the Colgate Global standards, to compete in emerging markets dominated by local players.
Herbal segment through launch of Herbal toothpaste
Pivotal in designing Consumer qualified products to enter new market segment
Efficiently executed Quality improvement programs for raw material across six manufacturing locations in India and Nepal
Margin improvement programs on all the major brands like Colgate Dental cream, toothpowder and Gel formulations thereby resulting in huge savings viz. common base Technology for to drive margins, new crystal structure development for Calcium carbonate, process improved for Triclosan manufacture, introduction of liquid form of Sodium lauryl sulphate, powder form of Sodium silicate, use of Natural calcium carbonate in liquid dentifrice
Developed self-preservation technique for liquid dentifrice named as Gold standard by Piscataway Research center
Merit of introducing Quality standards and Guidelines in India Technology center
Participated in designing new plant for toothpaste manufacture
Designing Specifications for Bureau of Indian Standards for Toothpaste, toothpowder, Sorbitol and Calcium carbonate
Filed one patent on Coated Natural Calcium Carbonate Oral Care Toothpowder Composition
Distinction of being committee member on three BIS Committees for PCD 19 for 4 years
Received Colgate YCMAD Award for innovation four times
Nominated team members and direct subordinates to win the award 19 times.
Sept ’1997-Jan1999 Searle India Ltd., Senior Manager (Pharmaceutical Development)
Notable Deliverables
·Instrumental in developing, reformulating & launching 12 products within fourteen months. Pivotal in developing
New products in area of conventional and specialized delivery system in therapeutic segment of cardiovascular, anti diabetic, gastrointestinal, anti-infective, neurological and biotechnology products.
Dosage forms like tablets, capsules, chewable, sustained release, dry syrups, liquid orals, suspensions using resin technology and injectables.
Played a key role in setting up a new department for international regulatory submissions, dossiers submitted for twenty molecules to contribute to export business.
July 1994 – Aug 1997 Lupin Laboratories Ltd as Senior Manager (Development)
Notable Deliverables
Led the effort in launching New Products Division , by developing & launching 8 new Herbal products through a new division of ethically promoted Natural products with 50 Sales personnel
Limited Phase III Clinicals for Anxiolytic and Appetite Stimulant formulation managed and coordinated through Clininvent
Periodontical Clinicals conducted and managed in Governmental dental College Mumbai
Claim support Clinicals on Ayurvedic Uterine tonic (U-Sedate) conducted at KEM Hospital Mumbai and Pune
Launched products like natural appetite stimulant, oral care, arthritis, rejuvenation, laxatives and digestive
Developed personal care products for export market, shampoos, hair oils, oral rinses, anti ageing cream all herbal/natural in nature
Successfully introduced bulk actives for exports, filed international patent on novel process for extraction of Hydroxyl Citric Acid from Garcinia.
Dec1991-June 1994 Ranbaxy Laboratories Ltd., as Product Development Manager
Notable Deliverables
Successfully developed and launched several conventional and specialty dosage forms as tablets, liquid oral, injectables, creams and led the team of product development scientists for India, Semi regulated markets of CIS , Indonesia , Malyasia and also Europe.
Managed and coordinated several BA/BE Studies for anti-infective in Ranbaxy and also Therapeutic Drug Monitoring Lab Mumbai.
Pivotal in supporting export market through regulatory submissions, product development and launch in the designated countries.
Significantly contributed in documentation for MCA approval.
March 1985-Nov 1991, Procter & Gamble India Ltd., as Research and Development Manager
Efficiently developed and launched indigenously developed Herbal products in OTC Category in
candy and syrup base
Actively participated in Clinical trials for OTC products (cough syrup)
Joining position Research Executive, promoted to Asstt. Manager, further promoted to Manager
Aug 80 -Dec ’84 Indian Institute of Technology, Benares Hindu University as Lecturer
Nov ’78-July ’80 Hamdard College of Pharmacy, University of Delhi as Lecturer
Academic Distinctions
Received Best Girl Student award in High School, University
Merit scholarship National Scholarship CSIR Scholarship –not availed
Memberships : Beureu of Indian Standards 2000-2004, IPA Life Member, CIM Member
regulatoryComments Off on New Q&A Document on the Visual Inspection of Parenterals available
Sep092016
The ECA’s Visual Inspection Group has developed a new document with answers to frequently asked questions. This new Q&A document is now available for download on the Group’s website. Read more about frequently asked questions in the visual inspection.
The Visual Inspection Group, an Interest Group of the ECA Foundation, has developed a new document with frequently asked questions. The new Q&A document, which was compiled by the Group’s Board, is now available for a free of charge download on the website.
For compiling the document the members of the Group were asked to provide their questions in February. These questions were then evaluated and answered by the Board Members.
The new document is structured as follows:
Manual Inspection
Automated Inspection
Qualification / Validation
Test sets
Re-Qualification
AQL-Tests
Defect Categorisation
Specific Products
Regulatory Affairs
Process Control / SPC
Some examples for the questions and the respective answers:
The grey portion of fully automatic control is often checked manually, to return not clearly or fully tested products back to the inspection process. Is it allowed to carry out this testing with the automated inspection machine?
From a GMP view, there are no restrictions. It is also important here that at the end a yield calculation and evaluation in the batch record appears. And there are also automated inspection systems that have already integrated the double inspection with multiple cameras.
In highly automated manufacturing lines for LVP flexible containers, the visual inspection process may/cannot comply to the standard visual inspection criteria e.g.: 5 sec inspection time, agitation of the container etc. Is this a compliance problem?
The requirements like 5 sec inspection time required by pharmacopoeias are addressing manually performed visual inspection. If the visual inspection is performed automatically, it is the company’s responsibility to ensure that the inspection via camera systems is as effective as a manual visual inspection via a validation (e.g. Knapp Test).
Should the AQL be inspected by QC or production
AQL manual inspection may be carried out by production staff (to avoid setting up a separate visual inspection team in QC) under a quality oversight or the quality unit. If performed by production operators, the AQL test should not be done by members of the team that was performing the 100 % visual inspection of the batch.
The Good Practice Guide “Visual Inspection of medicinal products for parenteral use”, was also revised. The new Version 3.0 will be introduced at the ECA Conference Particles in Parenterals in Barcelona, Spain, from 28-29 September 2016. All delegates of the conference will receive a free copy.
regulatoryComments Off on New Warning Letter of the FDA with the Focus on “Data Integrity”
Sep092016
The FDA has set the focus of its inspections on data integrity for quite some time already. The most recent Warning Letter addressed to a Chinese API manufacturer dated August 2016 clearly concentrates on the topic data integrity. Please find out more about the current FDA Warning Letter in this News.
Again, the focus of FDA’s Warning Letter for the Chinese API manufacturer Zhejiang Medicine Co. Ltd. dated 4th August 2016 is on the lack of data integrity. Among other things, records of activities were made not at the time when they have been performed. Moreover, original data have been deleted. A number of alarming findings were discovered in the course of the FDA inspection in June 2015.
The FDA is now expecting concrete measures (“Data Integrity Remediation”) from the company. For this, the FDA expressly recommended to retain qualified, external consultants. Among the measures to be taken:
A – A comprehensive investigation of the extent of incorrect data
1. An extensive plan for the execution of the investigation
2. Interviews of current and former employees to clarify the root cause of incorrect data
3. An assessment of the extent of data integrity deficits.
4. A comprehensive retrospective assessment of the performance of analytical testing.
B – A current risk assessment of the possible effects of the deficiencies identified on the quality of the medicinal products, up to the risk to patients!
C – A management strategy for the implementation of CAPA plans.
All in all, there were great concerns about the authenticity and reliability of the data produced in that company.





































 BESIFLOXACIN
BESIFLOXACIN








 DDD498
DDD498






































