Amtolmetin guacil,
ST-679, MED-15, Eufans
CAS 87344-06-7
UNII: 323A00CRO9,
Molecular Formula, C24-H24-N2-O5, Molecular Weight, 420.463,
Trade names: Amtoril®, Artricol®, Artromed®
US 4578481, US 6288241,
MEDOSAN RICERCA S.R.L. [IT/IT]; Via Cancelliera, 12 I-00040 Cecchina RM (IT) (For All Designated States Except US).
SIGMA-TAU INDUSTRIE FARMACEUTICHE RIUNITE S.P.A. [IT/IT]; Viale Shakespeare, 47 I-00144 Roma (IT)
Launched – 1993 ITALY, SIGMA TAU, Non-Opioid Analgesics FOR Treatment of Osteoarthritis, Treatment of Rheumatoid Arthritis,
- Originator sigma-tau SpA
- Class Amino acids; Antipyretics; Nonsteroidal anti-inflammatories; Pyrroles; Small molecules
- Mechanism of Action Cyclooxygenase inhibitors
-
Most Recent Events
- 01 Jun 1999 A meta-analysis has been added to the adverse events section
- 22 Jul 1995 Launched for Inflammation in Italy (PO)
Amtolmetin guacil is a NSAID which is a prodrug of tolmetin sodium.
Amtolmetin guacil is a nonacidic prodrug of tolmetin that has similar nonsteroidal antiinflammatory drug (NSAID) properties to those of Tolmetin with additional gastroprotective advantages. The term “nonsteroidal” is used to distinguish these drugs from steroids that have similar eicosanoid-depressing and antiinflammatory actions. Moreover, it possesses a more potent and long-lasting antiinflammatory activity than tolmetin and is marketed for the treatment of rheumatoid arthritis, osteoarthritis, and juvenile rheumatoid arthritis.
Background
Tolmetin sodium is an effective NSAID approved and marketed for the treatment of rheumatoid arthritis, osteoarthritis and juvenile rheumatoid arthritis. In humans, tolmetin sodium is absorbed rapidly with peak plasma levels observed 30 min after p.o. administration, but it is also eliminated rapidly with a mean plasma elimination t½ of approximately 1 hr. The preparation of slow release formulations or chemical modification of NSAIDs to form prodrugs has been suggested as a method to reduce the gastrotoxicity of these agents.
Amtolmetin guacil is a non-acidic prodrug of tolmetin, having similar NSAID properties like tolmetin with additional analgesic, antipyretic, and gastro protective properties. Amtolmetin is formed by amidation of tolmetin by glycine
Pharmacology
- Almost is absorbed on oral administration. It is concentrated maximum in internal the gastric wall, and highest concentration reached in 2 hours after administration.
- Amtolmetin guacil hydrolysed in to following metabolites Tolmetin, MED5 and Guiacol.
- Elimination will complete in 24 hours. Happens mostly with urine in shape of gluconides products (77%), faecal (7.5%).
- It is advised to take the drug on empty stomach.
- Permanent anti-inflammatory action is continued up to 72 hours, with single administration.
Mechanism of action
Amtolmetin guacil stimulates capsaicin receptors present on gastro intestinal walls, because of presence of vanillic moiety and also releases NO which is gastro protective. It also inhibits prostaglandin synthesis and cyclooxygenase (COX).

Structure of amtolmetin 1 and tolmetin 2.
26171-23-3 TOLMETIN FREE FORM
http://shodhganga.inflibnet.ac.in/bitstream/10603/2173/11/11_chapter%204.pdf
26171-23-3 TOLMETIN FREE FORM
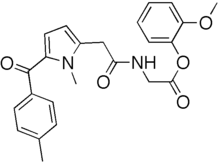
1-methyl-5-p-tolylpyrrole-2-acetic acid

Melting point 155-158 °C, IR (KBr, cm-1): 3205 (OH), 2958 (Aliphatic C-H), 1731 (Acid, C=O), 1700 (C=O), 1616 (C=C), 1267 (C-O); 1H NMR (CD3OD, 400 MHz): δ 7.63 ( d, J = 7.8 Hz, 2H), 7.27 (d, J = 7.8 Hz, 2H), 6.63 (d, J = 3.9 Hz, 1H), 6.11 (d, J = 4.3 Hz, 1H), 3.91 (s, 3H), 3.76 (s, 2H), 2.40 (s, 3H); MS (ESI): m/z calcd for C15H15NO3 (M + H): 258.11; found: (M + H) 257.9. (Fig. 4.12 – 4.14)




INNTERMEDIATE

1-methyl-5-p-toluoyl-2-acetamidoacetic acid
Melting point: 200-202° C. IR (KBr, cm-1): 3282 (NH), 3060 (OH), 1741 (Acid, C=O), 1637 (Amide, C=O), 1608 (C=C), 1178 (C-N); 1H NMR (CD3OD, 400 MHz): δ 7.64 ( dd, J =6.3 Hz, 1.9 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 6.65 (d, J = 3.9 Hz, 1H), 6.17 (d, J = 3.9 Hz, 1H), 3.92 (s, 3H), 3.73 (s, 2H), 3.30 (t, J =1.4 Hz, 2H), 2.41(s, 3H); MS (ESI): m/z calcd for C17H18N2O4 (M + H): 315.13; found: (M + H) 315. (Fig. 4.20 – 4.22)



SYNTHESIS

STUDENTS SOME COLOUR………………

1H and 13 C NMR PREDICT







SYNTHESIS
Amtolmetin guacil (CAS NO.: 87344-06-7), with its systematic name of N-((1-Methyl-5-p-toluoylpyrrol-2-yl)acetyl)glycine o-methoxyphenyl ester, could be produced through many synthetic methods.
Following is one of the synthesis routes: 1-Methyl-5-(4-methylbenzoyl)pyrrole-2-acetic acid (I) is condensed with glycine ethyl ester (II) in the presence of carbonyldiimidazole (CDI) and triethylamine in THF to afford the corresponding acetamidoacetate (III), which is hydrolyzed with NaOH in THF-water yielding 2-[2-[1-methyl-5-(4-methylbenzoyl)pyrrol-2-yl]acetamido]acetic acid (IV). Finally, this compound is esterified with 2-methoxyphenol (guayacol) (V) by means of CDI in hot THF.

PATENT
https://www.google.com/patents/WO1999033797A1?cl=tr
The present invention relates to a new crystalline form of 1- methyl-5-p-toluoylpyrrole-2-acetamidoacetic acid guaiacyl ester, a process for its preparation and to pharmaceutical compositions endowed with antiinflammatory, analgesic and antipyretic activity containing same.
The ester of 1-methyl-5-p-toluoylpyrrole-2-acetamidoacetic acid (hereinafter referred to as MED 15, form 1) is a known compound.
In fact, US Patent 4,882,349 discloses a class of N-mono- substituted and N,N-disubstituted amides of l-methyl-5-p- toluoylpyrrole-2-acetic acid (known as Tolmetin) endowed of anti- inflammatory, analgesic, antipyretic, antisecretive and antitussive properties.
US Patent 4,578,481 claims a specific compound, endowed with valuable pharmacological activity, encompassed in the above- mentioned class, precisely 1-methyl-5-p-toluoylpyrrole-2-acetamido- acetic acid guaiacyl ester (which is MED 15, form 1), and a process for its preparation.
The process disclosed in US 4,578,481 presents some drawbacks, since it is not easily applicable on industrial scale and gives low yields.
According to the above-mentioned process, Tolmetin was reacted with N,N’-carbonyldiimidazole in tetrahydrofuran (THF), and aminoacetic acid ethyl ester hydrochloride was added to the reaction mixture.
Following a complex series of washings in order to remove the unreacted starting compounds, and crystallisation from benzene/ cyclohexane, 1-methyl-5-p-toluoylpyrrole-2-acetamidoace-tic acid ethyl ester was obtained. This compound was subsequently transformed into the corresponding acid.
The acid was reacted with N,N’-carbonyldiimidazole obtaining the corresponding imidazolide, to which a solution of guaiacol in
THF was added.
From the reaction mixture, following several washings, neutralisation and crystallisation from benzene/ cyclohexane MED 15 form 1 was obtained.
The main physico-chemical characteristics of MED 15 form 1 are shown in table 1, left column.


The above mentioned process comprises the following steps:
(a) hydrolysing TOLMETIN 1 methyl ester with an alkaline hydroxide in a basic environment, obtaining TOLMETIN 2 alkaline salt;
(b) condensing 2 with isobutylchloroformate 3 obtaining the mixed anhydride 4;
(c) reacting 4 with glycine 5 obtaining 1-methyl-5-p-toluoylpyrrol-2- acetoamidacetic acid 6;
(d) condensing 6 with isobutylchloroformate 3 obtaining the mixed anhydride 7; and
(e) reacting the mixed anhydride 7 with guaiacol 8 obtaining 9 , MED 15, form 2.
The following non-limiting example illustrates the preparation of MED 15, form 2, according to the process of the present invention.
Preparation of 1-methyl-p-toluoylpirrol-2-acetoammidoacetic acid.
A mixture of 500 mL of toluene, 100 g of Tolmetin ethyl ester and 10 g of Terre deco in 1L flask, was heated to 70° C and maintained at this temperature for 20-30 min, under stirring. The mixture was then filtered on pre-heated buckner, and the solid phase washed with 50 mL of heated toluene. The discoloured toluene solution was transferred in a 2 L flask, 15 g of sodium hydroxide (97%) dissolved in 100 mL of water were added thereto.
The solution was heated at reflux temperature and refluxed for 1 hour. 22 mL of isobutyl alcohol were added to the solution which was heated at reflux temperature; water (about 120 mL) was removed completely with Marcusson’s apparatus arriving up to 104-105°C inner temperature.
To a suspension of Tolmetin sodium, cooled under nitrogen atmosphere to -5°C ± 2°C, 0.2 mL of N-methyl Morpholine were added. Maintaining the temperature at 0°C ± 3°C, 53 mL of isobutyl chloroformate were added dropwise in 5-10 min. After about 1 hour the suspension became fluid. Following 3 hours of reaction at 0°C + 3°C, over the glycine solution previously prepared, the mixed anhydride solution was added dropwise. The glycine solution was prepared in a flask containing 230 mL of demineralised water, 47 g of potassium hydrate (90%), cooling the solution to 20°C ± 2, adding 60 g of glycine, and again cooling to 10°C ± 2°C.
To the glycine solution, the mixed anhydride was added dropwise under stirring, in 5-10 min., maintaining the temperature at 20°C ± 2°C.
At the end of the addition, temperature was left to rise to room temperature, 1 hour later the reaction was complete. To the mixture 325 mL of demineralised water were added, the mixture was brought to pH 6.0 +2 using diluted (16%) hydrochloric acid (about 100 mL).
The temperature of the solution was brought to 73°C ±2°C and the pH adjusted to pH 5.0 ±0.2.
The separation of the two phases was made at hot temperature: the toluene phase was set aside for recovering acid-Tolmetin if any, the water phase was maintained at 73°C ±2°C and brought to pH 4.0 ±0.2 using diluted hydrochloric acid.
At the beginning of the precipitation the solution was slowly brought to pH 3.0 ±0.2 using diluted (16%) hydrochloric acid (100 mL).
The mixture was cooled to 15°C ±3°C and after 30 min. filtered. The solid cake was washed with 2×100 mL of demineralised water, the product was dried at 60°C under vacuum till constant weight. 100 g of 1-methyl-p-toluoylpirrol-2-acetoammidoacetic acid were obtained.
Preparation of MED 15, form 2
To a 2 L flask containing 730 mL of toluene, 100 g of dried compound of the above step were dissolved. To this solution 18.8 g of potassium hydrate (tit. 90%) in 65 mL of water were added.
The solution was dried maintaining the internal temperature at 95-100°C, and cooled to 55-60 °C. A solution of potassium hydrogen carbonate was then added and the resulting mixture was dried maintaining the internal temperature at 105°C ±2°C.
The mixture was cooled under nitrogen atmosphere to 5°C
±2°C, 24 mL of isobutyl alcohol and 0.3 mL of N-methyl morpholine were added thereto.
Maintaining the temperature at 10°C ±3°C, 47 mL of isobutyl-chloroformate were added dropwise in 5-10 minutes. The mixture was left to react for two hours at 10°C ±3°C obtaining an anhydride solution, which was added to a guaiacol solution previously prepared.
The guaiacol solution was prepared by loading in a 2L-flask 295 mL of water, 25 g of potassium hydrate (90%), and 0.3 g of sodium metabisulfite.
At the end of the loading the temperature was brought to 35-40°C.
The anhydride was added dropwise in 5- 10 min and the temperature was left to rise to room temperature.
The suspension was kept under stirring for 1 hour and brought to pH 6.0 ±0.5 with diluted hydrochloric acid. The suspension was heated to 70°C ± 5°C and maintained at pH 3-4 with diluted hydrochloric acid.
The phases were separated while hot. The aqueous phase was discharged, and to the organic phase, 250 mL of water were added.
Maintaining the temperature at 70 ±5°C the solution was brought to pH 8.0 ±0.5 with diluted sodium hydrate, the phases were separated while hot and the acqueous phase was discharged.
The organic phase was washed with 250 mL of water. At 70 ± 5°C the phases were separated. The toluene phase was then cleared with dicalite, filtered and left to crystallise.
The mixture was slowly cooled to 30°C – 35°C, the temperature was then brought to 10 ± 3°C and after 1 hour filtered, washed with toluene (2×100 mL).
The product was brought to dryness at 60°C under vacuum, thus giving 100 g of compound MED 15, form 2.
Theoretical yield: 133.7 g; Yield %: 74.8%.
PATENT
https://www.google.com/patents/WO2000032188A2?cl=un
PATENT
CN-100390144
PATENT
CN 1827597
Example 1: Steps:
Equipped with a trap, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water, drying the solution, when When the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 11.5g of potassium bicarbonate was added, and refluxed to remove water, until the internal temperature reaches 105 ± 2 ℃. The mixture was cooled to ice-water bath 5 ± 2 ℃, to which was added 24ml of isobutyl alcohol and 0.3ml N- methylmorpholine. The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel was added dropwise isobutylchloroformate 45.5ml (0.400mol), 10min addition was complete, so the mixture was 10 ± 3 ℃ 2hr reaction solution to obtain an acid anhydride, it has been prepared dropwise glycine guaiacol ester solution, 5-10min the addition was complete. Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 27g of potassium hydroxide (82%) and 0.3g of sodium metabisulfite, stirring to dissolve, the temperature was controlled at 10 ± 3 ℃, to which was added 82.7g (0.38mol) glycine guaiacol ester hydrochloride and prepared. Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, diluted with 16% hydrochloric acid to adjust the mixture to pH 6.0 ± 0.5. The suspension was heated to 70 ± 5 ℃, and then 16% diluted hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250ml of water, maintaining the temperature at 70 ± 5 ℃ with dilute (2N) sodium hydroxide solution to adjust the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded. With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 135.5 g, yield 90%. The crude product was recrystallized from acetone to give 1-methyl-5-acyl-2-acetyl-p-toluene amino acid ester of guaiacol boutique 127.9 g, yield 94.4%, mp128.7 ~ 131.9 ℃. Elemental analysis: C, 68.53%; H, 5.76%; N, 6.65%. IR spectrum (KBr tablet method): 3318,3142,2963,1778,1652,1626,1605,1500,1480,1456,13731255 and 1153cm-1.
Example 2: Procedure: equipped with a water separator, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water , drying the solution, when the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 11.5g of potassium bicarbonate was added, and refluxed to remove water, until the internal temperature reaches 105 ± 2 ℃. The mixture was cooled to ice-water bath 5 ± 2 ℃, to which was added 24ml of isobutyl alcohol and 0.3ml N- methylmorpholine. The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel dropwise isopropyl 46.5ml (0.41mol), 10-15min addition was complete, the mixture was allowed at 10 ± 3 ℃ reaction 2hr derived anhydride solution, it would have been prepared dropwise to glycine guaiacol ester solution, 5-10min the addition was complete. Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 27g of potassium hydroxide (82%) and 0.3g of sodium metabisulfite, stirring to dissolve, the temperature was controlled at 10 ± 3 ℃, to which was added 82.7g (0.38mol) glycine guaiacol ester hydrochloride and prepared. Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, diluted with 16% hydrochloric acid to adjust the mixture to pH 6.0 ± 0.5. The suspension was heated to 70 ± 5 ℃, and then 16% diluted hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250ml of water, maintaining the temperature at 70 ± 5 ℃ with dilute (2N) sodium hydroxide solution to adjust the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded. With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 138.5 g, yield 92%. The crude product was recrystallized from acetone to give 1-methyl-2-acyl-5-toluene acetaminophen acid ester guaiacol boutique 128.8 grams.
Example 3: equipped trap, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water, dried solution, when the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 10-12.5g potassium bicarbonate solution, refluxing was continued for removal of water, until the internal temperature reaches 105 ± 2 ℃. The mixture was cooled to ice-water bath 5 ± 2 ℃, added thereto 20-30ml of isobutyl alcohol 0.2-0.5mlN- methylmorpholine. The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel was added dropwise isobutylchloroformate 40.5-48.5ml, 10-15min addition was complete, so the mixture was 10 ± 3 ℃ 2hr reaction solution to obtain an acid anhydride, it has been prepared dropwise glycine guaiacol ester solution, 5-10min the addition was complete. Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 25-30g of potassium hydroxide (82%) or 15-17 grams of sodium hydroxide and sodium metabisulfite 0.2-0.5g or insurance powder, stirring to dissolve the temperature is controlled at 10 ± 3 ℃, to which is added 80-84g glycine guaiacol ester hydrochloride and prepared. Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, the mixture was adjusted with dilute hydrochloric acid to pH 6.0 ± 0.5. The suspension was heated to 70 ± 5 ℃, with dilute hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250-280ml of water, maintaining the temperature at 70 ± 5 ℃ , adjusted with dilute sodium hydroxide solution and the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded. With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 130-139 grams. The crude product was recrystallized from acetone to give 1-methyl-5-acyl-2-acetyl-p-toluene amino acid ester boutique guaiacol 120-129 grams.
PATENT
Indian Pat. Appl. (2010), IN 2008MU01617

DETAILED DESCRIPTION OF THE INVENTION
The present invention provides safe, environment friendly, economically viable and commercially feasible processes for the production of Amtolmetin guacil. There are two methods for the preparation of Amtolmetin guacil. The processes for the production of Amtolmetin guacil (I) comprise:
Method-1:
Step-A:- Treating 2-methoxy phenol of Formula VI with 2-(benzyloxycarbonylamino) acetic acid of Formula VII in the presence of an organic base and a condensing agent in chlorinated solvent to yield 2-methoxyphenyl-2- (benzyloxycarbonylamino) acetate of Formula V.
Step-B:- Acid addition salt of 2-methoxyphenyl -2-aminoacetate of Formula II may be prepared by treating 2-methoxyphenyl-2- (benzyloxycarbonylamino) acetate of Formula V with an acid and followed by crystallization in aprotic solvent.
7
Step-C):- l-methyl-5-p-toluoylpyrrole-2-acetic acid of Formula III is reacted with a condensing agent to form-activated moiety, which is reacted with acid addition salt of 2-methoxyphenyl -2-aminoacetate of Formula II in chlorinated solvent to produce Arntolmetin guacil of formula (I).
In a preferred embodiment of present invention, condensing agent used in step-A is selected from group consisting of dicyclohexylcarbodiimide, N, N’-carbonyl diimidazole, hydroxy benzotriazole. The most preferred condensing agent is Dicyclohexyl carbodiimide for the reaction.
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran, 1,4-dioxane, the most preferred solvent for the reaction methylene dichloride.
In another embodiment of the present invention, the reaction is performed in the presence of an organic base. The organic base is selected from the group consisting of trimethylamine, triethylamine, N-methyl morpholine, N-methylpyrrolidinone, 4-dimethyl Aminopyridine; the most preferred base is 4-dimethyl Aminopyridine.
In a preferred embodiment of present invention, the non-polar solvent used in step-B is selected from group consisting of ethers, hexanes, aromatic hydrocarbons and esters.
In another preferred embodiment of present invention, the most suitable solvents are esters.
In another preferred embodiment of present invention, condensing agent used in step-C is selected from group consisting of dicyclohexylcarbodiimide, N, N’-carbonyl diimidazole, hydroxy benzotriazole. The most preferred condensing agent is N, N’-carbonyl diimidazole for the conversion of the reaction.
8
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran, 1,4-dioxane, the most preferred solvent for the reaction methylene dichloride.
In yet another embodiment of the present invention, the reaction is performed at a temperature in the range of -20°C to 50°C. Most preferred temperature range for the reaction is (-) 10°C to 0°C.
Method-2:
Treating 2-(2-(I-methyl-5- (4-methylbenzoyl)-lH-pyrrol-2-yl) acetamido) acetic acid with 2-methoxy phenol in presence of condensing reagent and an organic base to obtain Amtolmetin guacil.
In a preferred embodiment of present invention, the condensing agent used is selected from group consisting of dicyclohexyicarbodiimide, hydroxy benzotriazole or a mixture thereof. The most preferred condensing agent is Dicvclohexyl carbodiimide for the aforementioned reaction.
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran. 1,4-dioxane, the most preferred solvent for the reaction is methylene dichloride.
In another embodiment of the present invention, the reaction is performed in the presence of an organic base. The organic base is selected from the group consisting of triethylamine, triethylamine, N-methyl morpholine, N-methylpyrrolidinone, 4-dimethyl Aminopyridine; the most preferred base is 4-dimethyl Aminopyridine.
9
In yet another embodiment of the present invention, the reaction is performed at a temperature in the range of -20°C to 50°C. Most preferred temperature range for the reaction is (-) 10°C to 0°C.
In another embodiment of present invention, crude amtolmetin guacil is directly purified using polar and non-polar solvent or a mixture thereof. The most preferred solvents are Isopropanol and toluene.
The following non-limiting examples illustrate specific embodiments of the present invention. They are, however, not intended to be limiting the scope of present invention in anyway.
Preparation of Amtolmetin guacil: Example-1;
Charged MDC (600 ml) and N-benzyloxycarbonyl glycine (100 gm) in a 2L-4NRBF under N2 atmosphere. Reaction mass was cooled down to -5°C. Added N, N’-dicyclohexylcarbodiimide solution (108.5 gm in 300 ml MDC) at-5°C to 0°C. Maintained temperature of reaction for 10 minutes at -5°C to 0°C. Added guaiacol solution (59.36 gm in 180 ml MDC) at -5°C to 0°C followed by addition of N, N-dimethyl aminopyridine (1 gm) at -5°C to 0°C. Monitored the reaction over TLC till the completion of reaction, while maintaining reaction at 0°C. Filtered the undissolved Dicyclohexyl urea and washed the solids with methylene dichloride (125 ml X 2). Collected filtrate and washing. Washed methylene dichloride with water (1000 ml X 2), lN-NaOH (500 ml X 2) and 1% HC1 solution (500 ml X 2), water (500 ml X 2) respectively. Organic methylene dichloride layer was dried over anhydrous sodium sulphate. Filtered sodium sulphate and collected methylene dichloride filtrate. Distilled out methylene dichloride under vacuum below 40°C to get oil. HPLC purity :> 90%
10
Added 33% HBr in acetic acid solution (262,5 gm) into reaction vessel at 25-30°C. Monitored the reaction over TLC till the completion of reaction, while maintaining the reaction at 25-30°C. Added ethyl acetate (1200 ml) slowly at 25-30°C after completion of reaction. Stirred the resultant slurry for 2.5 hours at 25-30°C for complete crystallization. Filtered the solids and washed it with ethyl acetate (200 ml). Dried solids at 50-55°C. Dry weight: 102 gm. HPLC Purity: >98%
Example-2:
Charged MDC (1400 ml) and N, N’-carbonyl di imidazole (69.34 gm) into a 3L-4NRBF under N2 atmosphere. Cooled it down to -15°C. Charged Tolmetin acid (100 gm) slowly into reaction vessel at -10° ± 5°C. Monitored the progress of reaction of over HPLC. After completion of reaction, charged slowly 2-methoxyphenyl-2- (benzyloxy carbonylamino) acetate hydrobromide salt (112.05 gm) at -10° ± 5°C.Monitored the reaction over HPLC. After completion of reaction, washed the organic layer with water (300 ml), 1% NaOH solution (100 ml) and water (300 ml X 2) respectively at 3-8°C. Treated organic layer with activated carbon (2.5 gm) and filtered over hyflow bed. Washed hyflow bed with methylene dichlonde (100 ml X 2). Distilled out methylene dichloride below 40°C under vacuum and stripped off traces with toluene (100 ml X 2) at 50-55°C. Charged toluene (600 ml) and Isopropanol (50ml). Heated the mass to 63-68°C. Stirred the clear solution at 63-68°C for 1 hour. Cooled it down slowly to 30°C followed by further cooling to 5°C. Stirred the resultant slurry for 3 hours at 0-5°C. Filtered solids and washed with toluene (100 ml X 2). Dried solids at 55-60°C under vacuum. Dry Weight: 130 gm. HPLC Purity: >99%
Example-3:
Charged MDC (333 liter) and 2-(2-(l-methyl-5- (4-methylbenzoyl)-lH-pyrrol-2-yl) acetamido) acetic acid (55.5 Kg) in reactor under N2 atmosphere at 25-30°C. Cool down reaction mass to -15 to -12°C. Added a freshly prepared solution of N, N’-dicyclohexyl
11
carbodiimide (47.39 Kg in 166.5 liter) slowly at -10° ± 5°C within 1 hour. Rinsed the addition funnel with MDC (55.5 liter) and added it to the reaction at -10° ± 5°C. Added guaiacol solution (24.14 Kg in 99.9 liter MDC) to the reaction mass at -10° ± 5°C within 1 hour. Rinsed the addition funnel with MDC (11.1 liter) and added to the reaction -10° ± 5°C. Charged N, N’-dimethyl aminopyridine (0.555 Kg) at -15°C. Maintained temperature of reaction mass at -10° ± 5°C for 3 hours. Monitored the reaction over TLC, After the completion of reaction, filtered the dicyclohexyl urea and washed the solids with MDC (55.5L X 2). Collected MDC filtrate and wash it with water (166.5 L X 2). Collected MDC layer and treated it with activated carbon (2.77 Kg) and filtered through sparkler. Washed the sparkler with MDC (111 L). Distilled out MDC below 40°C under vacuum and stripped off traces with toluene (55.5 L X 2) at 50-55°C. Charge toluene (333L) and Isopropanol (27.75 L). Heated reaction mass to 63-68°C to get a clear solution. Stirred the clear solution at 63-68°C for 1 hour. Cooled it down slowly to 30°C followed by further cooling to 20oC. Stirred the resultant slurry for 2 hours at 17-20°C. Filtered the solids and washed with toluene (55.5 L X 3). Dried the solids at 55-60°C under vacuum. Dry Weight: 48 Kg. HPLC Purity:>99%
PAPER
Synthesis and Process Optimization of Amtolmetin: An Antiinflammatory Agent†
Lekkala Amarnath Reddy‡, Saurya Chakraborty‡, Rodda Swapna‡, Dinesh Bhalerao‡, Golla China Malakondaiah‡, Mylavarapu Ravikumar‡, Abhijeet Kumar‡, Gade Srinivas Reddy‡, Jyothirmayi Naram‡, Namrata Dwivedi, Arnab Roy‡, Vurimidi Himabindu§, Bangaru Babu‡, Apurba Bhattacharya‡ and Rakeshwar Bandichhor*‡
Center of Excellence, Integrated Product Development, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Bachupalli, Qutubullapur, R. R. Dist. 500 072 Andhra Pradesh, India, and Center for Environment, Institute of Science and Technology, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad 500 072, India
Org. Process Res. Dev., 2010, 14 (2), pp 362–368
DOI: 10.1021/op900284w,
http://pubs.acs.org/doi/full/10.1021/op900284w
†DRL-IPD Communication number: IPDO IPM – 00202
, * Corresponding author. Telephone:
+91 40 44346430. Fax:
+91 40 44346164. E-mail:
[email protected].,
‡Dr. Reddy’s Laboratories Ltd.
, §Jawaharlal Nehru Technological University.
Abstract
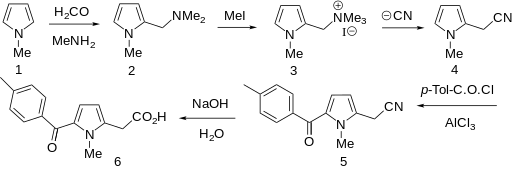
Efforts toward the synthesis and process optimization of amtolmetin guacil 1 are described. High-yielding electrophilic substitution followed by Wolf−Kishner reduction are the key features in the novel synthesis of tolmetin 2 which is an advanced intermediate of 1.
Amtolmetin Guacil
CAS Registry Number: 87344-06-7
CAS Name: N-[[1-Methyl-5-(4-methylbenzoyl)-1H-pyrrol-2-yl]acetyl]glycine 2-methoxyphenyl ester
Additional Names: N-[(1-methyl-5-p-toluoylpyrrol-2-yl)acetyl]glycine o-methoxyphenyl ester; 1-methyl-5-p-toluoylpyrrole-2-acetamidoacetic acid guaicil ester
Manufacturers’ Codes: ST-679; MED-15
Trademarks: Eufans (Sigma-Tau)
Molecular Formula: C24H24N2O5
Molecular Weight: 420.46
Percent Composition: C 68.56%, H 5.75%, N 6.66%, O 19.03%
Literature References: Ester prodrug of tolmetin, q.v. Prepn: A. Baglioni, BE 896018; idem, US 4578481 (1983, 1986 both to Sigma-Tau). Pharmacology: E. Arrigoni-Martelli, Drugs Exp. Clin. Res. 16, 63 (1990); A. Caruso et al., ibid. 18, 481 (1992). HPLC determn in plasma: A. Mancinelli et al., J. Chromatogr. 553, 81 (1991). Series of articles on pharmacokinetics and clinical trials:Clin. Ter. 142 (1 pt 2) 3-59 (1993).
Properties: Crystals from cyclohexane-benzene, mp 117-120°. Sol in common organic solvents. LD50 in male mice, rats (mg/kg): 1370, 1100 i.p.; >1500, 1450 orally (Baglioni).
Melting point: mp 117-120°
Toxicity data: LD50 in male mice, rats (mg/kg): 1370, 1100 i.p.; >1500, 1450 orally (Baglioni)
Therap-Cat: Analgesic; anti-inflammatory.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Arylacetic Acid Derivatives.
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////Amtolmetin guacil, ST-679, MED-15, Eufans, 87344-06-7, Amtoril®, Artricol®, Artromed®, амтолметин гуацил , أمتولمتين غواسيل , 呱氨托美丁
n1(c(ccc1CC(NCC(=O)Oc1c(cccc1)OC)=O)C(=O)c1ccc(cc1)C)C

































































tetracarboxylate_metallocarbene_stabilized_by_%CF%80C-Rh%E2%86%92%CF%80C%3DO_hyperconjugation..png)











![Image result for [3 + 2] Dipolar Cycloadditions](https://i0.wp.com/www.omicsonline.org/articles-images/theoretical-computational-science-Dipolar-Cycloaddition-Reaction-2-117-s002.png?w=695&ssl=1)