Entecavir
- Molecular FormulaC12H15N5O3
- Average mass277.279 Da
Baraclude (Entecavir) Film Coated Tablets & Oral Solution
Company: Bristol-Myers Squibb Pharmaceutical Co.
Application No.: 021797 & 021798
Approval Date: 03/29/2005

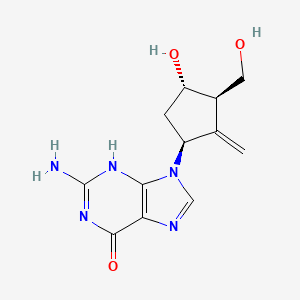
BARACLUDE® is the tradename for entecavir, a guanosine nucleoside analogue with selective activity against HBV. The chemical name for entecavir is 2-amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one, monohydrate. Its molecular formula is C12H15N5O3•H2O, which corresponds to a molecular weight of 295.3. Entecavir has the following structural formula:
 |
Entecavir is a white to off-white powder. It is slightly soluble in water (2.4 mg/mL), and the pH of the saturated solution in water is 7.9 at 25° C ± 0.5° C.
BARACLUDE film-coated tablets are available for oral administration in strengths of 0.5 mg and 1 mg of entecavir. BARACLUDE 0.5 mg and 1 mg film-coated tablets contain the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, crospovidone, povidone, and magnesium stearate. The tablet coating contains titanium dioxide, hypromellose, polyethylene glycol 400, polysorbate 80 (0.5 mg tablet only), and iron oxide red (1 mg tablet only). BARACLUDE Oral Solution is available for oral administration as a ready-to-use solution containing 0.05 mg of entecavir per milliliter. BARACLUDE Oral Solution contains the following inactive ingredients: maltitol, sodium citrate, citric acid, methylparaben, propylparaben, and orange flavor.


Entecavir is an oral antiviral drug used in the treatment of hepatitis B infection. It is marketed under the trade name Baraclude (BMS).
Entecavir is a guanine analogue that inhibits all three steps in the viral replication process, and the manufacturer claims that it is more efficacious than previous agents used to treat hepatitis B (lamivudine and adefovir). It was approved by the U.S. Food and Drug Administration (FDA) in March 2005.
For the treatment of chronic hepatitis B virus infection in adults with evidence of active viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
Entecavir (ETV), sold under the brand name Baraclude, is an antiviral medication used in the treatment of hepatitis B virus (HBV) infection.[1] In those with both HIV/AIDS and HBV antiretroviral medication should also be used.[1] Entecavir is taken by mouth as a tablet or solution.[1]
Common side effects include headache, nausea, high blood sugar, and decreased kidney function.[1] Severe side effects include enlargement of the liver, high blood lactate levels, and liver inflammation if the medication is stopped.[1] While there appears to be no harm from use during pregnancy, this use has not been well studied.[4] Entecavir is in the nucleoside reverse transcriptase inhibitors(NRTIs) family of medications.[1] It prevents the hepatitis B virus from multiplying by blocking reverse transcriptase.[1]
Entecavir was approved for medical use in 2005.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[5] In the United States as of 2015 it is not available as a generic medication.[6]The wholesale price is about 392 USD for a typical month supply as of 2016 in the United States.[7]
Medical uses
Entecavir is mainly used to treat chronic hepatitis B infection in adults and children 2 years and older with active viral replication and evidence of active disease with elevations in liver enzymes.[2] It is also used to prevent HBV reinfection after liver transplant[8] and to treat HIV patients infected with HBV. Entecavir is weakly active against HIV, but is not recommended for use in HIV-HBV co-infected patients without a fully suppressive anti-HIV regimen[9] as it may select for resistance to lamivudine and emtricitabine in HIV.[10]
The efficacy of entecavir has been studied in several randomized, double-blind, multicentre trials. Entecavir by mouth is effective and generally well tolerated treatment.[11]
Pregnancy and breastfeeding
It is considered pregnancy category C in the United States, and currently no adequate and well-controlled studies exist in pregnant women.[12]
Side effects
The majority of people who use entecavir have little to no side effects.[13] The most common side effects include headache, fatigue, dizziness, and nausea.[2] Less common effects include trouble sleeping and gastrointestinal symptoms such as sour stomach, diarrhea, and vomiting.[14]
Serious side effects from entecavir include lactic acidosis, liver problems, liver enlargement, and fat in the liver.[15]
Laboratory tests may show an increase in alanine transaminase (ALT), hematuria, glycosuria, and an increase in lipase.[16] Periodic monitoring of hepatic function and hematology are recommended.[2]
Mechanism of action
Entecavir is a nucleoside analog,[17] or more specifically, a deoxyguanosine analogue that belongs to a class of carbocyclic nucleosidesand inhibits reverse transcription, DNA replication and transcription in the viral replication process. Other nucleoside and nucleotide analogues include lamivudine, telbivudine, adefovir dipivoxil, and tenofovir.
Entecavir reduces the amount of HBV in the blood by reducing its ability to multiply and infect new cells.[18]
Administration
Entecavir is take by mouth as a tablet or solution. Doses are based on a person’s weight.[15] The solution is recommended for children more than 2 years old who weigh up to 30 kg. Entecavir is recommended on an empty stomach at least 2 hours before or after a meal, generally at the same time every day. It is not used in children less than 2 years old. Dose adjustments are also recommended for people with decreased kidney function.[15]
History
- 1992: SQ-34676 at Squibb as part of anti-herpes virus program[19]
- 1997: BMS 200475 developed at BMS pharmaceutical research institute as antiviral nucleoside analogue à Activity demonstrated against HBV, HSV-1, HCMV, VZV in cell lines & no or little activity against HIV or influenza[20]
- Superior activity observed against HBV pushed research towards BMS 200475, its base analogues and its enantiomer against HBV in HepG2.2.15 cell line[20]
- Comparison to other NAs, proven more selective potent inhibitor of HBV by virtue of being Guanine NA[21]
- 1998: Inhibition of hepadnaviral polymerases was demonstrated in vitro in comparison to a number of NAs-TP[22]
- Metabolic studies showed more efficient phosphorylation to triphosphate active form[23]
- 3-year treatment of woodchuck model of CHB à sustained antiviral efficacy and prolonged life spans without detectable emergence of resistance[24]
- Efficacy # LVD resistant HBV replication in vitro[25]
- Superior activity compared to LVD in vivo for both HBeAg+ & HBeAg− patients[26][27]
- Efficacy in LVD refractory CHB patients[28]
- Entecavir was approved by the U.S. FDA in March 2005.
Patent information
Bristol-Myers Squibb was the original patent holder for Baraclude, the brand name of entecavir in the US and Canada. The drug patent expiration for Baraclude was in 2015.[29][30]On August 26, 2014, Teva Pharmaceuticals USA gained FDA approval for generic equivalents of Baraclude 0.5 mg and 1 mg tablets;[31] Hetero Labs received such approval on August 21, 2015;[32] and Aurobindo Pharma on August 26, 2015.[33]
Chronic hepatitis B virus infection is one of the most severe liver diseases in morbidity and death rate in the worldwide range. At present, pharmaceuticals for treating chronic hepatitis B (CHB) virus infection are classified to interferon α and nucleoside/nucleotide analogue, i.e. Lamivudine and Adefovir. However, these pharmaceuticals can not meet needs for doctors and patients in treating chronic hepatitis B virus infection because of their respective limitation. Entecavir (ETV) is referred to as 2′-cyclopentyl deoxyguanosine (BMS2000475) which belongs to analogues of Guanine nucleotide and is phosphorylated to form an active triple phosphate in vivo. The triple phosphate of entecavir inhibits HBV polymerase by competition with 2′-deoxyguanosine-5′-triphosphate as a nature substrate of HBV polymerase, so as to achieve the purpose of effectively treating chronic hepatitis B virus infection and have strong anti-HBV effects. Entecavir, [1S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-hydroxymethyl]-2-methylenecyclopentyl]-6H-purin-6-one, monohydrate, and has the molecular formula of C12H15N5O3.H2O and the molecular weight of 295.3. Its structural formula is as follows:

Entecavir was successfully developed by Bristol-Myers Squibb Co. of USA first and the trademark of the product formulation is Baraclude™, including two types of formulations of tablet and oral solution having 0.5 mg and 1 mg of dosage. Chinese publication No. CN1310999 made by COLONNO, Richard, J. et al discloses a low amount of entecavir and uses of the composition containing entecavir in combination with other pharmaceutically active substances for treating hepatitis B virus infection, however, the entecavir is non-crystal. In addition, its oral formulations such as tablet and capsule are made by a boiling granulating process. The process is too complicated to control quality of products during humidity heat treatment even though ensuring uniform distribution of the active ingredients.
Entecavir, [1-S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one, is currently used for treating hepatitis B virus infection, whose structure is composed of a cyclopentane ring having purine, exomethylene, hydroxymethyl, and hydroxy substituents at the 1S-, 2-, 3R-, and 4S-positions, respectively. There have been conducted a number of studies to develop methods for preparing entecavir.
For example, U.S. Pat. No. 5,206,244 and WO 98/09964 disclose a method for preparing entecavir shown in Reaction Scheme 1:
The above method, however, has difficulties in that: i) the cyclopentadiene monomer must be maintained at a temperature lower than -30 °C in order to prevent its conversion to dicyclopentadiene; ii) residual sodium after the reaction as well as the sensitivity of the reaction toward moisture cause problems; iii) the process to obtain the intermediate of formula a) must be carried out at an extremely low temperature of below -70 °C in order to prevent the generation of isomers; iv) a decantation method is required when (-)-Ipc2BH (diisopinocampheylborane) is used for hydroboration; v) the process of the intermediate of formula a) does not proceed smoothly; and, vi) separation by column chromatography using CHP-20P resin is required to purify entecavir.
WO 2004/52310 and U.S. Pat. Publication No. 2005/0272932 disclose a method for preparing entecavir using the intermediate of formula (66), which is prepared as shown in Reaction Scheme 2:

The above preparation method of the intermediate of formula (66) must be carried out at an extremely low temperature of -70 °C or less, and the yield of the desired product in the optical resolution step is less than 50%.
PATENT
https://patents.google.com/patent/EP2382217B1

(3-4) Preparation of [1-S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purine-6-one (a compound of formula (1))
34 mg (0.115 mmol) of 4-(2-amino-6-chloro-purine-9-yl)-2-hydroxymethyl-3-methylene-cyclopentanol (a compound of formula (5)) obtained in (3-3) was added to 0.7 ml of 2N aqueous sodium hydroxide, and the resulting mixture was stirred. The solution thus obtained was heated to 72 °C and stirred for 3.5 hrs. After completion of the reaction, the resulting mixture was cooled to 0 °C, controlled to pH 6.3 by adding 2N aqueous hydrochloric acid and 1N aqueous hydrochloric acid, and condensed to obtain 24 mg of the title compound (yield: 70 %, purity: 99 %).
NMR(300MHz, DMSO-d6): δ 10.58 (s, 1H), 7.67 (s, 1H), 6.42 (s, 2H), 5.36 (t, 1H), 5.11 (s, 1H), 4.86 (d, 1H), 4.83 (t, 1H), 4.57 (s, 1H), 4.24 (s, 1H), 3.54 (t, 2H), 2.53(s, 1H), 2.27-2.18 (m, 1H), 2.08-2.01(m, 1H).
PAPER
https://www.sciencedirect.com/science/article/pii/S0040403911020144



PAPER
https://www.sciencedirect.com/science/article/pii/S0040402017313029


PAPER


Total Synthesis of Entecavir: A Robust Route for Pilot Production


A practical synthetic route for pilot production of entecavir is described. It is safe, robust, and scalable to kilogram scale. Starting from (S)-(+)-carvone, this synthetic route consists of a series of highly efficient reactions including a Favorskii rearrangement-elimination-epimerization sequence to establish the cyclopentene skeleton, the Baeyer–Villiger oxidation/rearrangement to afford the correct configuration of the secondary alcohol, and a directed homoallylic epoxidation followed by epoxide ring-opening to introduce the hydroxyl group suitable for the Mitsunobu reaction. In addition, the synthesis contains only four brief chromatographic purifications.
1: white crystalline solid; HRMS (m/z) calcd for C12H16N5O3 [M + H]+ 278.1253, found 278.1255; [α]D +27.2° [c 1.07, DMF/H2O (1:1)];
1H NMR (500 MHz, DMSO) δ 10.55 (s, 1H), 7.65 (s, 1H), 6.40 (s, 2H), 5.36 (dd, J = 10.3, 8.0 Hz, 1H), 5.10 (s, 1H), 4.85 (d, J = 3.1 Hz, 1H), 4.81 (t, J = 5.3 Hz, 1H), 4.56 (s, 1H), 4.23 (s, 1H), 3.54 (t, J = 6.1 Hz, 2H), 2.55–2.50 (m, 1H), 2.26–2.17 (m, 1H), 2.04 (dd, J = 12.5, 7.8 Hz, 1H);
13C NMR (126 MHz, DMSO) δ 156.8, 153.4, 151.4, 151.3, 135.9, 116.2, 109.2, 70.4, 63.0, 55.1, 54.1, 39.2.


Clips
EP 0481754; JP 1992282373; US 5206244, WO 9809964

The regioselective reaction of cyclopentadiene (I) and sodium or commercial sodium cyclopentadienide (II) with benzyl chloromethyl ether (III) by means of the chiral catalyst (-)-diisopinocampheylborane in THF, followed by hydroxylation with H2O2/NaOH, gives (1S-trans)-2-(benzyloxymethyl)-3-cyclopenten-1-ol (IV), which is regioselectively epoxidized with tert-butyl hydroperoxide and vanadyl acetylacetonate in 2,2,4-trimethylpentane, yielding [1S-(1alpha,2alpha,3beta,5alpha)-2-(benzyloxymethyl)-6-oxabicyclo[3.1.0]hexan-3-ol (V). The protection of (V) with benzyl bromide and NaH affords the corresponding ether (VI), which is condensed with 6-O-benzylguanine (VII) by means of LiH in DMF to give the guanine derivative (VIII). The protection of the amino group of (VIII) with 4-methoxyphenyl(diphenyl)chloromethane (IX), TEA and DMAP in dichloromethane gives intermediate (X), which is oxidized at the free hydroxyl group with methylphosphonic acid, DCC and oxalic acid in DMSO or Dess Martin periodinane in dichloromethane, yielding the cyclopentanone derivative (XI). The reaction of (XI) with (i) Zn/TiCl4/CH2Br2 complex in THF/CH2Cl2, (ii) activated Zn/PbCl2/CH2I2/TiCl4 in THF/CH2Cl2 (2), (iii) Nysted reagent/TiCl4 in THF/CH2Cl2 or (iv) Tebbe reagent in toluene affords the corresponding methylene derivative (XII), which is partially deprotected with 3N HCl in hot THF, providing the dibenzylated compound (XI). Finally, this compound is treated with BCl3 in dichloromethane
PAPER
Bioorg Med Chem Lett 1997,7(2),127
BMS-200475, a novel carbocyclic 2′-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro
BMS-200475, a novel carbocyclic analog of 2′-deoxyguanosine, is a potent inhibitor of hepatitis B virus in vitro (ED50 = 3 nM) with relatively low cytotoxicity (CC50 = 21–120 μM). A practical 10-step asymmetric synthesis was developed affording BMS-200475 in 18% overall chemical yield and >99% optical purity. The enantiomer of BMS-200475 as well as the adenine, thymine, and iodouracil analogs are much less active.
BMS-200475, a novel carbocyclic analog of 2′-deoxyguanosine, is a potent inhibitor of hepatitis B virus in vitro (ED50 = 3nM) with relatively low cytotoxicity (CC50 = 21–120 μM).

PATENT
https://patents.google.com/patent/US20140220120
Fourier transform infrared (FTIR) spectrogram: The range of wave numbers is measured by using the Nicolet NEXUS 670 FT-IR spectrometer with KBr pellet method, and the range of wave numbers is about 400 to 4000 cm−1. FIG. 3 is a Fourier transform infrared spectrogram of the sample. The infrared spectrogram shows that there are groups in the molecular structure of the sample, such as NH, NH2, HN—C═O, C═C, OH.

PAPER
Total Synthesis of Entecavir

Entecavir (BMS-200475) was synthesized from 4-trimethylsilyl-3-butyn-2-one and acrolein. The key features of its preparation are: (i) a stereoselective boron–aldol reaction to afford the acyclic carbon skeleton of the methylenecylopentane moiety; (ii) its cyclization by a Cp2TiCl-catalyzed intramolecular radical addition of an epoxide to an alkyne; and (iii) the coupling with a purine derivative by a Mitsunobu reaction.

2-Amino-9-((1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl)-1H-purin-6(9H)-one Monohydrate (1)
1 (2.102 g, 64% overall yield, 99.47% HPLC purity) with a 6.7% water content (as determined by Karl Fischer titration). Mp 248 °C. [α]D25 +35.0 (c 0.4, H2O). IR (ATR): 3445, 3361, 3296, 3175, 3113, 2951, 2858, 2626, 1709 cm–1.
1H NMR (DMSO-d6, 400 MHz) δ: 10.59 (s, 1H), 7.66 (s, 1H), 6.42 (bs, 2H), 5.36 (ddt, J = 10.6, 7.8, 2.7 Hz, 1H), 5.10 (dd, J = 2.7, 2.2 Hz, 1H), 4.87 (d, J = 3.1 Hz, 1H), 4.84 (t, J = 5.3 Hz, 1H), 4.56 (t, J = 2.4 Hz, 1H), 4.23 (m, 1H), 3.53 (m, 2H), 2.52 (m, 1H), 2.22 (ddd, J = 12.6, 10.8, 4.6 Hz, 1H), 2.04 (ddt, J = 12.6, 7.7, 1.9 Hz, 1H).
13C NMR (DMSO-d6, 101 MHz) δ: 156.9, 153.5, 151.5, 151.3, 136.0, 116.2, 109.3, 70.4, 63.1, 55.2, 54.1, 39.2. HRMS (ESI): m/z calcd for C12H16N5O3+ [M + H]+ 278.1253; found 278.1262.
PATENTS
PublicationPublication DateTitle
References
- “Entecavir”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
- “Baraclude (entecavir) Tablets for Oral Use & Oral Solution. U.S. Full Prescribing Information. Archived 2014-02-22 at the Wayback Machine.” Bristol-Myers Squibb Company, 2005. Revised December 2013.
- The Merck Index (14th ed.). 2006. p. 613. ISBN 978-0-911910-00-1.
- “Entecavir (Baraclude) Use During Pregnancy”. www.drugs.com. Archived from the original on 7 November 2016. Retrieved 6 December 2016.
- “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 76. ISBN 9781284057560.
- “NADAC as of 2016-11-30 | Data.Medicaid.gov”. Centers for Medicare and Medicaid Services. Archived from the original on 30 November 2016. Retrieved 6 December 2016.
- Fung, J; Cheung, C; Chan, SC; et al. (2011). “Entecavir Monotherapy is Effective in Suppressing Hepatitis B Virus After Liver Transplantation”. Gastroenterology. 141 (4): 1212–9. doi:10.1053/j.gastro.2011.06.083. PMID 21762659.
- “Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents” (PDF). Panel on Antiretroviral Guidelines for Adults and Adolescents. Archived (PDF) from the original on 1 November 2016. Retrieved 15 March 2015.
- McMahon, Moira (21 June 2007). “The Anti-Hepatitis B Drug Entecavir Inhibits HIV-1 Replication and Can Select HIV-1 Variants Resistant to Antiretroviral Drugs”. N Engl J Med. 356 (25): 2614–2621. doi:10.1056/NEJMoa067710. PMC 3069686
 . PMID 17582071. Archived from the original on 2 April 2015. Retrieved 15 March 2015.
. PMID 17582071. Archived from the original on 2 April 2015. Retrieved 15 March 2015. - Scott, LJ; Keating, GM (2009). “Entecavir”. Drugs. 69 (8): 1003–1033. doi:10.2165/00003495-200969080-00005. Archived from the original on 2011-10-08.
- Jump up^ “Entecavir (Baraclude) Use During Pregnancy”. www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-07.
- Jump up^ “Entecavir: Indications, Side Effects, Warnings – Drugs.com”. www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-10.
- Jump up^ “Entecavir Side Effects in Detail – Drugs.com”. www.drugs.com. Archived from the original on 2016-11-10. Retrieved 2016-11-10.
- ^ Jump up to:a b c “DailyMed – BARACLUDE- entecavir tablet, film coated BARACLUDE- entecavir solution”. dailymed.nlm.nih.gov. Archived from the original on 2016-11-08. Retrieved 2016-11-09.
- Jump up^ “DailyMed – BARACLUDE- entecavir tablet, film coated BARACLUDE- entecavir solution”. dailymed.nlm.nih.gov. Archived from the original on 2016-11-09. Retrieved 2016-11-10.
- Jump up^ Sims KA, Woodland AM (December 2006). “Entecavir: a new nucleoside analog for the treatment of chronic hepatitis B infection”. Pharmacotherapy. 26 (12): 1745–57. doi:10.1592/phco.26.12.1745. PMID 17125436.[permanent dead link]

- Jump up^ “Entecavir: Indications, Side Effects, Warnings – Drugs.com”. www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-07.
- Jump up^ Slusarchyk, W. A., A. K. Field, J. A. Greytok, P. Taunk, A. V. Tooumari, M. G. Young, and R. Zahler. 4-Hydroxy-3-(hydroxymethyl)-2-methylcyclopentyl purines and pyrimidines, a novel class of anti-herpesvirus agents. Abstract from the Fifth International Conference on Antiviral Research. Antivir Res 1992.17(Suppl. 1):98
- ^ Jump up to:a b Bisacchi, G. S.; Chao, S. T.; Bachard, C.; Daris, J. P.; Innaimo, S. F.; Jacobs, J. A.; Kocy, O.; Lapointe, P.; Martel, A.; Merchant, Z.; Slusarchyk, W. A.; Sundeen, J. E.; Young, M. G.; Colonno, R.; Zahler, R. (1997). “BMS-200475, a novel carbocyclic 29-deoxyguanosine analog with potent and selective antihepatitis B virus activity in vitro”. Bioorg. Med. Chem. Lett. 7: 127–132. doi:10.1016/s0960-894x(96)00594-x.
- Jump up^ Innaimo, S F; Seifer, M; Bisacchi, G S; Standring, D N; Zahler, R; Colonno, R J (1997). “Identification of BMS-200475 as a Potent and Selective Inhibitor of Hepatitis B Virus. Antimicrob”. Agents Chemother. 41 (7): 1444–1448.
- Jump up^ Seifer, M.; Hamatake, R. K.; Colonno, R. J.; Standring, D. N. (1998). “In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob”. Agents Chemother. 42: 3200–3208.
- Jump up^ Yamanaka, G.; Wilson, T.; Innaimo, S.; Bisacchi, G. S.; Egli, P.; Rinehart, J. K.; Zahler, R.; Colonno, R. J. (1999). “Metabolic studies on BMS-200475, a new antiviral compound active against hepatitis B virus. Antimicrob”. Agents Chemother. 43: 190–193.
- Jump up^ Colonno, R. J.; Genovesi, E. V.; Medina, I.; Lamb, L.; Durham, S. K.; Huang, M. L.; Corey, L.; Littlejohn, M.; Locarnini, S.; Tennant, B. C.; Rose, B.; Clark, J. M. (2001). “Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection”. J. Infect. Dis. 184: 1236–1245. doi:10.1086/324003.
- Jump up^ Levine, S.; Hernandez, D.; Yamanaka, G.; Zhang, S.; Rose, R.; Weinheimer, S.; Colonno, R. J. (2002). “Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob”. Agents Chemother. 46: 2525–2532. doi:10.1128/aac.46.8.2525-2532.2002.
- Jump up^ Chang, T. T. (2006). “A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B”. N. Engl. J. Med. 354: 1001–1010. doi:10.1056/nejmoa051285.
- Jump up^ Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, Goodman Z, DeHertogh D, Wilber R, Zink RC, Cross A, Colonno R, Fernandes L (9 March 2006). “Entecavir versus Lamivudine for Patients with HBeAg-Negative Chronic Hepatitis B”. The New England Journal of Medicine. 354 (10): 1011–20. doi:10.1056/NEJMoa051287. PMID 16525138.
- Jump up^ Sherman, M.; Yurdaydin, C.; Sollano, J.; Silva, M.; Liaw, Y. F.; Cianciara, J.; Boron-Kaczmarska, A.; Martin, P.; Goodman, Z.; Colonno, R. J.; Cross, A.; Denisky, G.; Kreter, B.; Hindes, R. (2006). “Entecavir for the treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B”. Gastroenterology. 130: 2039–2049. doi:10.1053/j.gastro.2006.04.007.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. www.accessdata.fda.gov. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. Orange Book. Patent and Exclusivity for: N021798. Archived from the original on 15 November 2016. Retrieved 14 November 2016.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “202122.”. Archived from the original on 22 December 2015. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “205740.”. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “206217.”. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ɛnˈtɛkəvɪər/ en-TEK-a-vir or en-TE-ka-veer |
| Trade names | Baraclude[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605028 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
by mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | n/a (≥70)[2] |
| Protein binding | 13% (in vitro) |
| Metabolism | negligible/nil |
| Biological half-life | 128–149 hours |
| Excretion | Renal 62–73% |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.111.234 |
| Chemical and physical data | |
| Formula | C12H15N5O3 |
| Molar mass | 277.279 g/mol |
| 3D model (JSmol) | |
| Melting point | 220 °C (428 °F) value applies to entecavir monohydrate and is a minimum value[3] |
///////////////Entecavir, энтекавир , إينتيكافير , 恩替卡韦 , BMS-200475, SQ-200475, エンテカビル,
NC1=NC(=O)C2=C(N1)N(C=N2)[C@H]1C[C@H](O)[C@@H](CO)C1=C
NMR PREDICT
1H NMR AND 13C NMR



13C PREDICT VALUES

























 Open Access
Open Access