
PITAVASTATIN, LIVALO, Itavastatin calcium, Nisvastatin, NKS-104, NK-104,
CAS REGISTRY NUMBER
rotation is +
alpha(D20) +6.8° (c 1.74, CHCl3)
ALSO
Bioorganic and Medicinal Chemistry Letters, 1999 , VOL 9, 20 pg. 2977 – 2982…….alpha(D20) +23.1° (c 1.0, acn/water(1:))
Helvetica Chimica Acta, 2007 , vol. 90, 6 pg. 1069 – 1081…alpha(D20) +22.9° (c 1.0, acn/water)
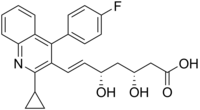
(3R,5S,6E)-7-[2-cyclopropyl-4-(4-fluorophenyl)quinolin-3-yl]-3,5-dihydroxyhept-6-enoic acid
Pitavastatin a lipid-lowering agent that belongs to the statin class of medications for treatment of dyslipidemia. It is also used for primary and secondary prevention of cardiovascular disease. FDA approved in Aug 3, 2009.
2-C25-H23-FN-O4.Ca, 881.01

Statin drugs are currently the most therapeutically effective drugs available for reducing the level of Low density lipoprotein (LDL) in the blood stream of a patient at risk for cardiovascular disease. A high level of LDL in the
bloodstream has been linked to the formationof coronary lesions which obstruct the flow of blood and can rupture and promote thrombosis. It is well known that inhibitors against HMG CoA reductase which is rate limiting enzyme for cholesterol biosynthesis have been clinically proved to be potentially useful anti-hyperlipoproteinemic agents
and they are considered very effective curative and preventive for coronary artery sclerosis or atherosclerosis .
Pitavastatin calcium was discovered by Nissan Chemical Industries Limited Japan and developedfurther by Kowa Pharmaceuticals Tokyo Japan is a novel member of the medication class of statins.
LIVALO (pitavastatin) is an inhibitor of HMG-CoA reductase. It is a synthetic lipid-lowering agent for oral administration.
The chemical name for pitavastatin is (+)monocalcium bis{(3R, 5S, 6E)-7-[2-cyclopropyl-4-(4-fluorophenyl)-3-quinolyl]-3,5dihydroxy-6-heptenoate}. The structural formula is:
 |
The empirical formula for pitavastatin is C50H46CaF2N2O8 and the molecular weight is 880.98. Pitavastatin is odorless and occurs as white to pale-yellow powder. It is freely soluble in pyridine, chloroform, dilute hydrochloric acid, and tetrahydrofuran, soluble in ethylene glycol, sparingly soluble in octanol, slightly soluble in methanol, very slightly soluble in water or ethanol, and practically insoluble in acetonitrile or diethyl ether. Pitavastatin is hygroscopic and slightly unstable in light.
Each film-coated tablet of LIVALO contains 1.045 mg, 2.09 mg, or 4.18 mg of pitavastatin calcium, which is equivalent to 1 mg, 2 mg, or 4 mg, respectively of free base and the following inactive ingredients: lactose monohydrate, low substituted hydroxypropylcellulose, hypromellose, magnesium aluminometasilicate, magnesium stearate, and film coating containing the following inactive ingredients: hypromellose, titanium dioxide, triethyl citrate, and colloidal anhydrous silica.

Pitavastatin (usually as a calcium salt) is a member of the blood cholesterol loweringmedication class of statins,[1] marketed in the United States under the trade nameLivalo. Like other statins, it is an inhibitor of HMG-CoA reductase, the enzyme that catalyses the first step of cholesterol synthesis. It has been available in Japan since 2003, and is being marketed under licence in South Korea and in India.[2] It is likely that pitavastatin will be approved for use in hypercholesterolaemia (elevated levels of cholesterol in the blood) and for the prevention of cardiovascular disease outside South and Southeast Asia as well.[3] In the US, it received FDA approval in 2009.[4]
Pitavastatin is used to lower serum levels of total cholesterol, LDL-C, apolipoprotein B, and triglycerides, and raise levels of HDL-C for the treatment of dyslipidemia.
Like the other statins, pitavastatin is indicated for hypercholesterolaemia (elevated cholesterol) and for the prevention of cardiovascular disease. A 2009 study showed that pitavastatin increased HDL cholesterol (24.6%), especially in patients with HDL lower than 40 mg/dl, in addition to greatly reducing LDL cholesterol (–31.3%).[5] As a consequence, pitavastatin is most likely to be appropriate for patients with metabolic syndrome with high LDL, low HDL and diabetes mellitus.
Common statin-related side effects (headaches, stomach upset, abnormal liver function tests and muscle cramps) were similar to other statins. However, pitavastatin seems to lead to fewer muscle side effects than certain statins that are lipid-soluble, as a result of the fact that pitavastatin is water-soluble (as is pravastatin, for example).[6] One study found that coenzyme Q10 was not reduced as much as with certain other statins (though this is unlikely given the inherent chemistry of the HMG-CoA reductase pathway that all statin drugs inhibit).[3][7]
Hyperuricemia or increased levels of serum uric acid have been reported with pitavastatin.[8]

Most statins are metabolised in part by one or more hepatic cytochrome P450enzymes, leading to an increased potential for drug interactions and problems with certain foods (such as grapefruit juice). Pitavastatin appears to be a substrate ofCYP2C9, and not CYP3A4 (which is a common source of interactions in other statins). As a result, pitavastatin is less likely to interact with drugs that are metabolized via CYP3A4, which might be important for elderly patients who need to take multiple medicines.[3]
Pitavastatin (previously known as itavastatin, itabavastin, nisvastatin, NK-104 or NKS-104) was discovered in Japan by Nissan Chemical Industries and developed further byKowa Pharmaceuticals, Tokyo.[3] Pitavastatin was approved for use in the United States by the FDA on 08/03/2009 under the trade name Livalo. Pitavastatin has been also approved by the Medicines and Healthcare products Regulatory Agency (MHRA) in UK on 17 August 2010.
- Kajinami, K; Takekoshi, N; Saito, Y (2003). “Pitavastatin: efficacy and safety profiles of a novel synthetic HMG-CoA reductase inhibitor”.Cardiovascular drug reviews 21 (3): 199–215. PMID 12931254. edit
- Zydus Cadila launches pitavastatin in India
- Mukhtar, R. Y. A.; Reid, J.; Reckless, J. P. D. (2005). “Pitavastatin”. International Journal of Clinical Practice 59 (2): 239–252.doi:10.1111/j.1742-1241.2005.00461.x. PMID 15854203. edit
- The Seventh Statin; Pitavastatin
- http://www.ncbi.nlm.nih.gov/pubmed/19907105
- ScienceDaily (11 May 2013). “Alternative Cholesterol-Lowering Drug for Patients Who Can’t Tolerate Statins”. ScienceDaily.
- Clin Pharmacol Ther. 2008 May;83(5):731-9. Epub 2007 Oct 24. Comparison of effects of pitavastatin and atorvastatin on plasma coenzyme Q10 in heterozygous familial hypercholesterolemia: results from a crossover study. Kawashiri MA, Nohara A, Tada H, Mori M, Tsuchida M, Katsuda S, Inazu A, Kobayashi J, Koizumi J, Mabuchi H, Yamagishi M.
- Ogata, N.; Fujimori, S.; Oka, Y.; Kaneko, K. (2010). “Effects of Three Strong Statins (Atorvastatin, Pitavastatin, and Rosuvastatin) on Serum Uric Acid Levels in Dyslipidemic Patients”. Nucleosides, Nucleotides and Nucleic Acids 29 (4–6): 321.doi:10.1080/15257771003741323. edit


|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 7,022,713 | 2009-08-03 | 2024-02-19 |
| United States | 6,465,477 | 2009-08-03 | 2016-12-20 |
| United States | 5,856,336 | 2009-08-03 | 2016-01-05 |
| United States | 5,854,259 | 2009-08-03 | 2015-12-29 |
| United States | 5,753,675 | 2009-08-03 | 2015-05-19 |
JP 1993310700, JP 1994025092
Tetrahedron Lett 1993, 34, 51, 8263-6.
Bioorg Med Chem2001, 9, (10): 2727
Drugs Fut1998, 23, (8) :847-859
Bull Chem Soc Jpn1995, 68, (1) :364-72
Tetrahedron Asymmetry1993, 4, (2) :201-4
Tetrahedron Lett1993, 34, (51) :8267-70
A Endo; J. Med. Chem. 1985, 28, 401.
AM Gotta; LC Smith; IXth International Symp. Drugs Affecting Lipid Metabolism, 1986, 30- 31
Y Fujikawa, Nissan Chemical Industries Ltd, EP304063 (A3), 1989.
S Ahmed; CS Madsen; PD Stein; J. Med. Chem. 2008, 51, 2722-2733.
K Turner; Org. Process. Res. Dev, 2004, 8, 823-833.
Z Casar; M Steinbocher; J Kosmrlj; J. Org. Chem. 2010, 75(19), 6681-6684.
KL Baumann; Tetrahedron Letters, 1992, 33, 2283-2284.
N Miyachi; Y Yanagawa; H Iwasaki; Tetrahedron Lett. 1993, 34, 8267-8270.
T Minami; K Takahashi; T Hiyama; Tetrahedron Lett. 1993, 34, 3, 513-516.
DA Evans; AH Hoveyda; J. Org. Chem. 1990, 55, 5190-5192.
J Castorer; LA Sorbera; PA Leeson; Drugs Fut. 23(8), 1998, 847-859.
T Hiyama; K Takahashi; T Minami; Bull. Chem. Soc. Jpn. 1995, 68, 364-372.
MS Reddy; M Bairy; K Reddy; Oriental Journal of Chemistry. 2007, 23, 559-564.
RN Moore; G Bigam; JK Chan; AM Hogg; JC Vederas; J. Am. Chem. Soc. 1985, 107 3694-3701.
DS Johnson; JJ Li; Art of Drug Synthesis, John Wiley & Sons, New Jersey, 2007, 177-181.
MT Stone; Organic Lett. 2011, 13, 2326-2329.
SR Manne, SR Maramreddy, WO2007132482 (A2), 2007.
SD Dwivedi, DJ Patel, AP Shah, Cadila Healthcare Ltd, US0022102 (A1), 2012.
……………………………………………………..

……………………………

The reaction of 1 (R) ,7,7-trimethylbicyclo [2.2.1] heptan-2-one (I) with 1 -naphthylmagnesium bromide (II) gives the tertiary alcohol (III), which by reaction with SOCl2 and then with NaHCO3 yields 2 – (1-naphthyl) -1 (R) ,7,7-trimethylbicyclo [2.2.1] heptene (IV ). Hydroboration of (IV) with BH3 followed by oxidation with H2O2 affords 4 (S) ,7,7-trimethyl-3exo-(1-naphthyl) bicyclo [2.2.1] heptan-2exo-ol (V), which is submitted to transesterification with methyl acetoacetate (VI) and dimethyl-aminopyridine (DMAP) to give the corresponding ester (VII). The condensation of (VII) with N-methoxy-N-methyl-3-[2-cyclopropyl-4-( 4-fluorophenyl) quinolin-3-yl] -2 (E)-propenamide (VIII) by means of NaH yields the corresponding chiral 3,5-dioxoheptenoic acid ester (IX), which is selectively reduced first with diisobutylaluminum hy-dride acid (DIBAL) and then with diethylmethoxyborane and sodium borohydride affording the 3 (R), 5 (S)-dihydroxyheptenoic ester (X). Finally, this compound is saponified with NaOH and treated with acetic acid / sodium acetate. The intermediate amide (VIII ) is obtained by condensation of 2-cyclopropyl-4-(4-fluorophenyl) quinoline-3-carbaldehyde (XI) with N-methoxy-N-methylacetamide (XII) by means of butyllithium to the hydroxy propionamide (XIII), which is then dehydrated with methanesulfonyl chloride and triethylamine in the usual way).
…………………

A systematic chiral synthesis of NK-104 and its enantiomer (X) has been reported: The oxidation of the already known 2-cyclopropyl-4-(4-fluorophenyl)quinoline-3-methanol (I) with DMSO, P2O5 and triethylamine gives the corresponding aldehyde (II), which is condensed with diethyl cyanomethylphosphonate by means of NaOH in toluene yielding the propenenitrile (III). The reduction of (III) with DIBAL affords the unsaturated aldehyde (IV), which is condensed with ethyl acetoacetate by means of NaH and n-BuLi to provide the 3-oxo-5-hydroxy-6-heptenoic acid ethyl ester derivative (V). The highly syn stereoselective reduction of (V) by means of diethylmethoxyborane and NaBH4 yields the desired syn racemic mixture of erythro-beta,delta-dihydroxyesters (VII), which is submitted to optical resolution with chiral (+)-alpha-methylbenzylamine [(+)-MBA] to obtain NK-104 free acid (VIII), which is finally treated with NaOH and CaCl2. The enantiomer of NK-104 has been obtained by optical resolution of the racemic mixture (VII) with (-)-alpha-methylbenzylamine to obtain the enantiomeric free acid (IX), which is treated with NaOH and CaCl2 as before.
Fujikawa, Y.; Suzuki, M.; Iwasaki, H.; Kitahara, M.; Sakashita, M.; Sakoda, R.;. Synthesis and biological evaluations of quinolone-based HMG-CoA reductase inhibitors Bioorg Med Chem 2001, 9 , 10, 2727
………


ADDITIONAL UPDATED INFO
Pitavastatin calcium is a novel member of the medication class of statins. Marketed in the United States under the trade name Livalo, it is like other statin drugs an inhibitor of HMG-CoA reductase, the enzyme that catalyses the first step of cholesterol synthesis. It is likely that pitavastatin will be approved for use in hypercholesterolaemia (elevated levels of cholesterol in the blood) and for the prevention of cardiovascular disease outside South and Southeast Asia as well.
Pitavastatin calcium is chemically known as (3R,5S)-7-[2-cyclopropyl-4-(4-fluorophenyl)quinolin-3-yl]-3,5-dihydroxy-6(E)-heptenoic acid calcium salt having the formula IA is known in the literature.

Pitavastatin is a synthetic lipid-lowering agent that acts as an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme a (HMG-CoA) reductase (HMG-CoA Reductase inhibitor). This enzyme catalyzes the conversions of HMG-CoA to mevalonate, inhibitors are commonly referred to as “statins”. Statins are therapeutically effective drugs used for reducing low density lipoprotein (LDL) particle concentration in the blood stream of patients at risk for cardiovascular disease. Pitavastatin is used in the treatment of hyperchloesterolemia and mixed dyslipidemia.
Pitavastatin calcium has recently been developed as a new chemically synthesized and powerful statin by Kowa Company Ltd, Japan. On the basis of reported data, the potency of Pitavastatin is dose-dependent and appears to be equivalent to that of Atorvastatin. This new statin is safe and well tolerated in the treatment of patients with hypercholesterolaemia. Significant interactions with a number of other commonly used drugs can be considered to be extremely low.
Pitavastatin was disclosed for the first time in US patents US 4,761,419, US 5,01 1 ,930 and US 5,753,675. The process disclosed in these patents for the preparation of Pitavastatin is as shown below:

wherein R is hydrogen or protecting group.
US 5,284,953 discloses a process for the preparation of Pitavastatin calcium, which employs optically active a-methylbenzylamine as a resoluting agent.
The above processes are economically not viable, as resolution is carried out in final stage.
US 6,835,838 B2 discloses a process for the preparation of Pitavastatin calcium, which is as shown below:


However, it has been observed that the above process of lactonization results in ~10- 15% of unreacted Pitavastatin ethyl ester and therefore results in low yield. Further, -10% of Pitavastatin acid results during the above lactonization process and therefore does not produce a single product which is required to keep adequate control for an intermediate through specifications to have consistently better quality of the finished product.
Processes for the preparation of Pitavastatin are described in EP-A-0304063 and EP-A-1099694 and in the publications by N. Miyachi et al. in Tetrahedron Letters (1993) vol. 34, pages 8267-8270 and by K. Takahashi et al. in Bull. Chem. Soc. Japan (1995) Vol. 68, 2649-2656. These publications describe the synthesis of Pitavastatin in great detail but do not describe the hemi-calcium salt of Pitavastatin. The publications by L A. Sorbera et al. in Drugs of the Future (1998) vol. 23, pages 847-859 and by M. Suzuki at al. in Bioorganic & Medicinal Chemistry Letters (1999) vol. 9, pages 2977-2982 describe Pitavastatin calcium, however, a precise procedure for its preparation is not given. A full synthetic procedure for the preparation of Pitavastatin calcium is described in EP-A-0520406. In the process described in this patent Pitavastatin calcium is obtained by precipitation from an aqueous solution as a white crystalline material with a melting point of 190-192° C.
US20090182008 A1 discloses polymorphic form A, B, C, D, E, and F, and the amorphous form of Pitavastatin Calcium salt (2:1). In particular, crystalline Form A having water content from about. 5% to about 15% and process for its preparation are disclosed.
US20090176987 A1 also discloses polymorphic form crystal form A of Pitavastatin Calcium which contains from 5 to 15% of water and which shows, in its X-ray powder diffraction as measured by using CuKa radiation, a peak having a relative intensity of more than 25% at a diffraction angle (20) of 30.16°.
WO2007/132482 A1 discloses a novel process for the preparation of Pitavastatin Calcium by condensing bromide salt of formula-3 with aldehyde compound of formula-4 to obtain olefinic compound of formula-5 and converting olefinic compound to Pitavastatin Calcium via organic amine salt for purification.
Pitavastatin and its process were disclosed in U.S. Pat. No. 5,753,675.
Pitavastatin calcium and its process were disclosed in U.S. Pat. No. 5,856,336. PCT publication no. WO 2004/072040 (herein after referred to ‘040 patent) disclosed crystalline polymorph A, polymorph B, polymorph C, polymorph D, polymorph E, polymorph F and amorphous form of pitavastatin calcium
-
Synthesis of pitavastatin via cross-coupling reaction is disclosed inTetrahedron Lett. 1993, 34, 8263-8266, and in Tetrahedron Lett. 1993, 34, 8267-8270.
-
A method for the preparation of pitavastatin via epichlorohydrin is described in Tetrahedron: Asymmetry 1993, 4, 201-204.
-
Synthesis of pitavastatin heterocycle and pitavastatin molecule assembly via aldol condensation reaction is disclosed in Bioorg. Med. Chem. Lett. 1999, 9, 2977-2982, and Bioorg. Med. Chem. 2001, 9, 2727-2743:


-
PCT application WO 2003/064382 describes a method for preparation of pitavastatin by asymmetric aldol reaction, in which titanium complex is used as a catalyst.
-
HWE route to pitavastatin by utilization of 3-formyl substituted pitavastatin heterocycle is disclosed in Helv. Chim. Acta 2007, 90, 1069-1081:
-
Methods for preparation of pitavastatin heterocycle derivatives are described in Bull. Chem. Soc. Jpn. 1995, 68, 364-372, Heterocycles 1999, 50, 479-483, Lett. Org. Chem. 2006, 3, 289-291, and in Org. Biomol. Chem. 2006, 4, 104-110, as well as in the international patent applications WO 95/11898 and WO 2004/041787
-
WO 95/11898 and Bull. Chem. Soc. Jpn. 1995, 68, 364-372 disclose synthesis of PTVBR from PTVOH with PBr3:




WO 1995/1 1898 Al discloses a process for the preparation of Pitavastatin, which is as shown below:

wherein Y represents P+RnRi2Ri3Hal“ or P(W)Ri4R15; R9a, R% and R]0 are protecting groups each of Rn, Rj2> R^, Ri4 and R15 which are independent of one another, is optionally substituted alkyl or optionally substituted aryl group; R14 and Rj5 together form a 5- or 6-membered ring; Hal is chlorine, bromine or iodine; and W is O or S.
The above process results in 2-5% of Cis isomer of Pitavastatin which requires further purification and therefore results poor yield.
US 6,875,867 B2 discloses a process for the preparation of Pitavastatin arginine salt, which is as shown below:

Saponification / Base

During the above process Trifluoroacetic acid or hydrochloric acid is used to break the acetonide and the Pitavastatin ester formed is converted in situ to its corresponding alkali salt by treating with base, such as sodium hydroxide.
US20090182008 A1 discloses polymorphic form A, B, C, D, E, and F, and the amorphous form of Pitavastatin Calcium salt (2:1). In particular, crystalline Form A having water content from about. 5% to about 15% and process for its preparation are disclosed.
……………………………………..
nmr
http://scholarsresearchlibrary.com/dpl-vol4-iss5/DPL-2012-4-5-1553-1557.pdf
calcium bis-(E)-3,5-dihydroxy-7-[4’-(4’’-flurophenyl)-2’-
cyclopropyl-quinoline-3-yl]-hept-6-enoate , pitavastatin calcium
Melting Point: 207 degC;
IR υmax (KBr) cm-1: 3366 (OH), 2911, 1603 (C=O), 1567 (C=N), 1513 (C=C),
1488 (C-H), 1416 (C-H), 1313, 1275, 1221 (C-O-C), 1158, 1065 (C-H), 972, 843, 763.
1H-NMR (500MHz, DMSO-d6):
δ 1.01 (m, 2H), 1.09 (m, 1H), 1.19 (m, 2H), 1.41 (m, 1H),
1.98 (dd, 1H, J1 =8.5,
J2 =15.5Hz), 2.11(d, 1H, J1 =3.0, J2 =15.5Hz), 2.50 (m, 2H),
3.66 (m, 1H), 4.13 (m, 1H), 4.95 (s, 1H), 5.58 (dd, 1H,
J1 =5.5, J2 =10.5Hz), 6.49 (d, 1H, J = 16.0Hz),
7.35 (m, 6H), 7.59 (m, 1H, J = 7.0Hz), 7.83 (d, 1H, J =8.5Hz).
13CNMR & DEPT (125.76MHz, DMSO-d6):
δ 11.12(CH2, C-17), 11.23(CH2,C-18), 15.80(CH2, C-16), 44.29(CH2,
C-22), 44.61(CH2, C-24), 66.61(C-O, C-23),
69.34(C-O,C-21),115.53(C=C, C-20), 15.62(CH), 115.79(CH),
123.59(CH), 126.07(C=C, C-19),
128.79(CH),129.20(CH),130.07(CH), 32.30(CH),
132.56(CH), 133.51(C),
142.60(C), 144.09(C), 146.37(C),
161.02(C), 163.00(C), 179.13(C=O, C-25).
ESI-MS: m/z (%) 318 (100), 274 (23), 423 (13), 422 (M+, 70); EI calcd for C25H24FNO4, 421.461; found, 422.220
(M+).
…………………

………………


























































































