CASOPITANT
Tachykinin NK1 Antagonists
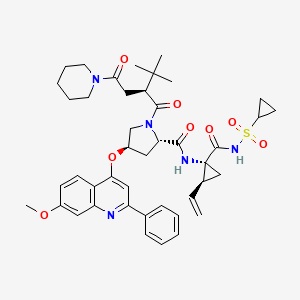
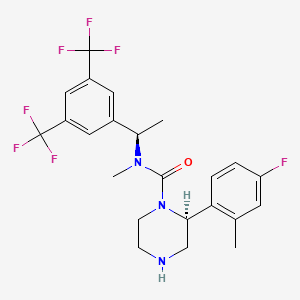
(2S,4S)-4-(4-Acetyl-1-piperazinyl)-N-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-2-methylphenyl)-N-methyl-1-piperidinecarboxamide
4-(S)-(4-Acetyl-piperazin-1-yl)-2- (R)-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid, [1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide
414910-30-8 MESYLATE
414910-27-3 (free base)
679769
GW-679769
GW-679769B
| MF C30H35F7N4O2.CH4O3S MESYLATE | |
| Molecular Weight | 712.719 |
Casopitant (trade names Rezonic (US), Zunrisa (EU)) is an neurokinin 1 (NK1) receptor antagonist undergoing research for the treatment of chemotherapy-induced nausea and vomiting (CINV).[1] It is currently under development by GlaxoSmithKline (GSK).
In July 2008, the company filed a marketing authorisation application with the European Medicines Agency. The application was withdrawn in September 2009 because GSK decided that further safety assessment was necessary.[2] Casopitant mesylate, a tachykinin NK1 receptor antagonist, had been filed for approval in the U.S. and the E.U. by GlaxoSmithKline for the prophylaxis of chemotherapy-induced nausea/vomiting.
In 2009 the company discontinued the development of the drug candidate for this indication. An MAA had also been filed for the treatment of postoperative nausea and vomiting, and in 2009 the application was withdrawn by the company.
Additional phase II clinical trials were ongoing at GlaxoSmithKline for the treatment of depression, anxiety, sleep disorders, fibromyalgia and overactive bladder, however, no recent developments have been reported for these indications.
- Lohr L (2008). “Chemotherapy-induced nausea and vomiting”. Cancer J 14 (2): 85–93.doi:10.1097/PPO.0b013e31816a0f07. PMID 18391612.
- “GlaxoSmithKline withdraws its marketing authorisation application for Zunrisa”. London: EMEA. 13 October 2009. Retrieved 21 December 2009
- Casopitant mesilate
Drugs Fut 2008, 33(9): 737 - WO 2002032867
- WO 2008046882
- Development of a control strategy for a defluorinated analogue in the manufacturing process of casopitant mesylate
Org Process Res Dev 2010, 14(4): 832 NMR FREE BASE, MESYLATE - WO 2006061233
- WO 2004091616
- US20040014770 ENTRY 1B MP MESYLATE 243
- Tetrahedron, 2010 , vol. 66, 26 p. 4769 – 4774 NMR FREE BASE
- Journal of Medicinal Chemistry, 2011 , vol. 54, 4 p. 1071 – 1079 NMR MESYLATE
| WO2006061233A1 * | Dec 7, 2005 | Jun 15, 2006 | Glaxo Group Ltd | The use of medicament 4-(s)-(4-acetyl-piperazin-1-yl)-2-(r)-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid, [1-(r)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamide |
| WO2001044200A2 * | Dec 14, 2000 | Jun 21, 2001 | David J Blythin | Selective neurokinin antagonists |
| WO2002010141A1 * | Jul 25, 2001 | Feb 7, 2002 | Michael Kirk Ahlijanian | Imidazole derivatives |
| WO2002032867A1 * | Oct 12, 2001 | Apr 25, 2002 | Giuseppe Alvaro | Chemical compounds |
| US20020123491 * | Dec 14, 2000 | Sep 5, 2002 | Neng-Yang Shih | Selective neurokinin antagonists |
| US20030064980 * | Jun 6, 2002 | Apr 3, 2003 | Neng-Yang Shih | Selective neurokinin antagonists |
| US20030144270 * | Nov 12, 2002 | Jul 31, 2003 | Schering Corporation | NK1 antagonists |

Casopitant (Rezonic, Zunrisa, casopitant mesylate, GW-679769, 679769, CAS #414910-27-3), 4-(4-Acetyl-piperazin-1-yl)-2-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid [1-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide, is a NK-1 receptor antagonist.
Casopitant is under investigation for the treatment of emesis, nausea, drug-induced nausea, chemotherapy-induced nausea and vomiting, post-operative nausea and vomiting, sleep disorders, anxiety disorders, depressive disorders, overactive bladder, and myalgia (Drug Report for Casopitant, Thomson Investigational Drug Database (Sep. 15, 2008); Reddy et al., Supportive Cancer Therapy 2006, 3(3), 140-142; and WO 2006/061233).
Casopitant has also shown promise in treating disorders of the central nervous system, tinitis, and sexual dysfunction (WO 2006/061233).
compound may be of value in the treatment of Sexual dysfunctions including Sexual Desire Disorders such as Hypoactive Sexual Desire Disorder and Sexual Aversion Disorder sexual arousal disorders such as Female Sexual Arousal Disorder and Male Erectile Disorder orgasmic disorders such as Female Orgasmic Disorder, Male Orgasmic Disorder and Premature Ejaculation sexual pain disorder such as Dyspareunia and Vaginismus, Sexual Dysfunction Not Otherwise Specified; paraphilias such as Exhibitionism, Fetishism, Frotteurism, Pedophilia, Sexual Masochism, Sexual Sadism Transvestic Fetishism, Voyeurism and Paraphilia Not Otherwise Specified gender identity disorders such as Gender Identity Disorder in Children and Gender Identity Disorder in Adolescents or Adults and Sexual Disorder Not Otherwise Specified.

Casopitant is subject to CYP3A4-mediated oxidative metabolism at the 3-carbon of the piperazine ring to form a hydroxylated metabolite which may be further oxidized to the corresponding 3-oxo metabolite (Minthorn et al, Drug Metab. Disp., 2008, 36(9), 1846-1852). Adverse effects associated with casopitantadministration include: neutropenia, nausea, hiccups, headache, constipation, dizziness, pruritis, alopecia, and fatigue.
Overactive bladder is a term for a syndrome that encompasses urinary frequency, with or without urge incontinence, generally but not necessarily combined with pollacisuria and nocturia. Overactive bladder is also characterised by involuntary detrusor contractions which are either triggered by provocation or occur spontaneously. If the detrusor hyperactivity observed is based on neurological causes (e. g. Parkinson’s disease, apoplexy, some forms of multiple sclerosis, spinal cord injury or the cross section of the bone marrow) it is known as neurogenic detrusor hyperactivity. If no clear cause can be detected this is known as idiopathic detrusor hyperactivity. In addition, detrusor hyperactivity may be associated with anatomical changes in the lower urinary tract, for example, in patients with bladder outlet obstruction (an enlargement of the prostate gland in males)
International patent application WO 02/32867 describes novel piperidine derivatives. A 0 particular preferred compound described therein is 4-(S)-(4-Acetyl-piperazin-1-yl)-2-(R)- (4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid
…………………………………………………………………
CASOPITANT MESYLATE
EXAMPLE 4
[0330] 4-(S)-(4-Acetyl-piperazin-1-yl)-2-(R)-(4-fluoro-2-methyl-phenyl)-piperidine-1-Carboxylic Acid, [1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamide Methanesulphonate
[0331] A solution of intermediate 4a (7.7 g) in acetonitrile (177 mL) was added to a solution of 1-acetyl-piperazine (3.9 g) in acetonitrile (17.7 mL) followed by sodium triacetoxyborohydride (6.4 g) under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 24 hours and then quenched with a saturated sodium hydrogen carbonate (23.1 mL) and water (61.6 mL). The resulting solution was concentrated in vacuo, then AcOEt (208 mL) was added; the layers were separated and the aqueous layer was back-extracted with further AcOEt (2×77 mL). The collected organic phases were washed with brine (2×118 mL), dried and concentrated in vacuo to give the crude mixture of syn and anti diastereomers (nearly 1:1) as a white foam (9.5 g).
[0332] A solution of this intermediate in THF (85.4 mL) was added to a solution of methansulfonic acid (0.890 mL) in THF (6.1 mL) at r.t. After seeding, the desired syn diastereomer started to precipitate. The resulting suspension was stirred for 3 hours at 0° C. and then filtered under a nitrogen atmosphere. The resulting cake was washed with cold THF (15.4 mL) and dried in vacuo at +20° C. for 48 hours to give the title compound as a white solid (4.44 g).
[0333] NMR (d6-DMSO): δ (ppm) 9.52 (bs, 1H); 7.99 (bs, 1H); 7.68 (bs, 2H); 7.23 (m, 1H); 6.95 (dd, 1H); 6.82 (m, 1H); 5.31 (q, 1H); 4.45 (bd, 1H); 4.20 (dd, 1H); 3.99 (bd, 1H); 3.65-3.25 (bm, 5H); 3.17 (m, 1H); 2.96 (m, 1H); 2.88-2.79 (m+m, 2H); 2.73 (s, 3H); 2.36 (s, 3H); 2.30 (s, 3H); 2.13-2.09 (bd+bd, 2H); 2.01 (s, 3H); 1.89-1.73 (m+m, 2H); 1.46 (d, 3H).
[0334] m.p 243.0° C.
[0335] The compound is isolated in a crystalline form.
intermediate 4a is needed for above syn, ignore 4b
[0168] Intermediate 4
[0169] 2-(R)-(4-Fluoro-2-methyl-phenyl)-4-oxo-piperidine-1-Carboxylic Acid, [1-(R)-3,5-bis-trifluoromethyl-phenyl)ethyl]-Methylamide (4a) and 2-(S)-(4-Fluoro-2-methyl-phenyl)-4-oxo-piperidine-1-Carboxylic Acid, [1-(R)-3,5-bis-trifluoromethyl-phenyl)-ethyl]-Methylamide (4b) Method A
[0170] A solution of triphosgene (147 mg) dissolved in dry DCM (5 mL) was added drop-wise to a solution of intermediate 2 (250 mg) and DIPEA (860 μL) in dry DCM (15 mL) previously cooled to 0° C. under a nitrogen atmosphere. After 2 hours, [1-(R)-3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride (503 mg) and DIPEA (320 μL) in dry acetonitrile (20 mL) were added and the mixture was heated to 70° C. for 16 hours. Further [1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride (170 mg) and DIPEA (100 μL) were added and the mixture was stirred at 70° C. for further 4 hours. Next, the mixture was allowed to cool to r.t., taken up with AcOEt (30 mL), washed with a 1N hydrochloric acid cold solution (3×15 mL) and brine (2×10 mL). The organic layer was dried and concentrated in vacuo to a residue, which was purified by flash chromatography (CH/AcOEt 8:2) to give:
[0171] 1. intermediate 4a (230 mg) as a white foam,
[0172] 2. intermediate 4b (231 mg) as a white foam. …………….ignore
[0173] Intermediate 4a
[0174] NMR (d6-DMSO): δ (ppm) 7.98 (bs, 1H); 7.77 (bs, 2H); 7.24 (dd, 1H); 6.97 (dd, 1H); 6.89 (m, 1H); 5.24 (t, 1H); 5.14 (q, 1H); 3.61 (m, 1H); 3.55 (m, 1H); 2.71 (m, 2H); 2.56 (s, 3H); 2.50 (m, 2H); 2.26 (s, 3H); 1.57 (d, 3H).
[0175] Intermediate 4b
[0176] NMR (d6-DMSO): δ (ppm) 7.96 (bs, 1H); 7.75 (bs, 2H); 7.24 (dd, 1H); 6.98 (dd, 1H); 6.93 (dt, 1H); 5.29 (q, 1H); 5.24 (t, 1H); 3.56 (m, 1H); 3.48 (m, 1H); 2.70 (s, 3H); 2.50 (m, 4H); 2.26 (s, 3H); 1.54 (d, 3H). …….. ignore
[0177] Intermediate 4a
[0178] Method B
[0179] A saturated sodium hydrogen carbonate solution (324 mL) was added to a solution of intermediate 9 (21.6 g) in AcOEt (324 mL) and the resulting mixture was vigorously stirred for 15 minutes. The aqueous layer was back-extracted with further AcOEt (216 mL) and the combined organic extracts were dried and concentrated in vacuo to give intermediate 8 as a yellow oil, which was treated with TEA (19 mL) and AcOEt (114 mL). The solution obtained was added drop-wise over 40 minutes to a solution of triphosgene (8 g) in AcOEt (64 mL) previously cooled to 0° C. under a nitrogen atmosphere, maintaining the temperature between 0° C. and 8° C.
[0180] After stirring for 1 hours at 0° C. and for 3 hours at 20° C., [1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride (29.7 g), AcOEt (190 mL) and TEA (38 mL) were added to the reaction mixture which was then heated to reflux for 16 hours.
[0181] The solution was washed with 10% sodium hydroxide solution (180 mL), 1% hydrochloric acid solution (4×150 mL), water (3×180 mL) and brine (180 mL). The organic layer was dried and concentrated in vacuo to a residue, which was purified through a silica pad (CH/AcOEt 9:1) to give the title compound (21.5 g) as a brown thick oil.
[0182] NMR (d6-DMSO): 6 (ppm) 7.97-7.77 (bs+bs, 3H); 7.24 (dd, 1H); 6.97 (dd, 1H); 6.88 (td, 1H); 5.24 (m, 1H); 5.14 (q, 1H); 3.58 (m, 2H); 2.7 (m, 2H); 2.56 (s, 3H); 2.49 (m, 2H); 2.26 (s, 3H); 1.57 (d, 3H).
intermediate 2
[0152] Intermediate 2
[0153] 2-(4-Fluoro-2-methyl-phenyl)-piperidine-4-one
[0154] Method A
[0155] 2-Methyl-4-fluoro-benzaldehyde (4 g) was added to a solution of 4-aminobutan-2-one ethylene acetal (3.8 g) in dry benzene (50 mL) and the solution was stirred at r.t. under a nitrogen atmosphere. After 1 hour the mixture was heated at reflux for 16 hours and then allowed to cool to r.t. This solution was slowly added to a refluxing solution of p-toluensulphonic acid (10.6 g) in dry benzene (50 mL) previously refluxed for 1 hour with a Dean-Stark apparatus. After 3.5 hours the crude solution was cooled and made basic with a saturated potassium carbonate solution and taken up with AcOEt (50 mL). The aqueous phase was extracted with AcOEt (3×50 mL) and Et2O (2×50 mL). The organic layer was dried and concentrated in vacuo to a yellow thick oil as residue (7.23 g). A portion of the crude mixture (3 g) was dissolved in a 6N hydrochloric acid solution (20 mL) and stirred at 60° C. for 16 hours. The solution was basified with solid potassium carbonate and extracted with DCM (5×50 mL). The combined organic phases were washed with brine (50 mL), dried and concentrated in vacuo to give the title compound (2.5 g) as a thick yellow oil.
[0156] Method B
[0157] L-selectride (1M solution in dry THF, 210 mL) was added drop-wise, over 80 minutes, to a solution of intermediate 1 (50 g) in dry THF (1065 mL) previously cooled to −72° C. under a nitrogen atmosphere. After 45 minutes, 2% sodium hydrogen carbonate solution (994 mL) was added drop-wise and the solution was extracted with AcOEt (3×994 mL). The combined organic phases were washed with water (284 mL) and brine (568 mL). The organic phase was dried and concentrated in vacuo to get 1-benzyloxycarbonyl-2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one as a pale yellow thick oil (94 g) which was used as a crude.
[0158] This material (94 g) was dissolved in AcOEt (710 mL), then 10% Pd/C (30.5 g) was added under a nitrogen atmosphere. The slurry was hydrogenated at 1 atmosphere for 30 minutes. The mixture was filtered through Celite and the organic phase was concentrated in vacuo to give the crude 2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one as a yellow oil. This material was dissolved in AcOEt (518 mL) at r.t. and racemic camphorsulphonic acid (48.3 g) was added. The mixture was stirred at r.t for 18 hours, then the solid was filtered off, washed with AcOEt (2×50 mL) and dried in vacuo for 18 hours to give 2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one, 10-camphorsulfonic acid salt as a pale yellow solid (68.5 g). (M.p.: 167-169° C.-NMR (d6-DMSO): 6 (ppm) 9.43 (bs, 1H); 9.23 (bs, 1H); 7.66 (dd, 1H); 7.19 (m, 2H); 4.97 (bd, 1H); 3.6 (m, 2H); 2.87 (m, 3H); 2.66 (m, 1H); 2.53 (m, 2H); 2.37 (s+d, 4H); 2.22 (m, 1H); 1.93 (t, 1H); 1.8 (m, 2H); 1.26 (m, 2H); 1.03 (s, 3H); 0.73 (s, 3H).
[0159] This material (68.5 g) was suspended in AcOEt (480 mL) and stirred with a saturated sodium hydrogen carbonate (274 mL). The organic layer was separated and washed with further water (274 mL). The organic phase was dried and concentrated in vacuo to give the title compound (31 g) as a yellow-orange oil.
[0160] NMR (d6-DMSO): 6 (ppm) 7.49 (dd, 1H); 7.00 (m, 2H); 3.97 (dd, 1H); 3.27 (m, 1H); 2.82 (dt, 1H); 2.72 (bm, 1H); 2.47 (m, 1H); 2.40 (m, 1H); 2.29 (s, 3H); 2.25 (dt, 1H); 2.18 (m, 1H).
[0161] MS (ES/+): m/z=208 [MH]+.
intermediate 9
[0220] Intermediate 9
[0221] 2-(R)-(4-Fluoro-2-methyl-phenyl)-piperidin-4-one Mandelic Acid.
[0222] A solution of L-(+)-mandelic acid (22.6 g) in AcOEt (308 mL) was added to a solution of intermediate 2 (31 g) in AcOEt (308 mL). Then isopropanol (616 mL) was added and the solution was concentrated in vacuo to 274 mL. The solution was then cooled to 0° C. and further cold isopropanol (96 mL) was added. The thick precipitate was stirred under nitrogen for 5 hours at 0° C., then filtered and washed with cold Et2O (250 mL) to give the title compound as a pale yellow solid (20.3 g).
[0223] M.p.: 82-85° C.
[0224] NMR (d6-DMSO): δ (ppm) 7.51 (dd, 1H); 7.40 (m, 2H); 7.32 (m, 2H); 7.26 (m, 1H); 7.0 (m, 2H); 4.95 (s, 1H); 4.04 (dd, 1H); 3.31 (m, 1H); 2.88 (m, 1H); 2.49-2.2 (m, 4H); 2.29 (s, 3H).
[0225] Chiral HPLC: HP 1100 HPLC system; column Chiralcel OD-H, 25 cm×4.6 mm; mobile phase: n-hexane/isopropanol 95:5+1% diethylamine; flow: 1.3 ml/min; detection: 240/215 nm; retention time 12.07 minutes.
………………….
NMR
mesylate
http://pubs.acs.org/doi/abs/10.1021/op100150b

1H NMR (600 MHz, DMSO-d6): 9.57 (br s, 1H), 7.99 (br s, 1H), 7.68 (br s, 2H), 7.23 (m, 1H), 6.95 (dd, 1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99 (m, 1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96 (m, 1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30 (s, 3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73 (m, 1H), 1.46 (d, 3H), MS: m/z 617 [MH]+, as free base.
……………
http://pubs.acs.org/doi/full/10.1021/op100209c
 11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.

……………………………….
http://pubs.acs.org/doi/full/10.1021/jm1013264?prevSearch=casopitant&searchHistoryKey=
(2R,4S)-1′-acetyl-N-{(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4,4′-bipiperidine-1-carboxamide methanesulfonate salt 16a (casopitant)
nmr mesylate
1H NMR (600 MHz, DMSO-d6): 9.57 (bs, 1H), 7.99 (bs, 1H), 7.68 (bs, 2H), 7.23 (m, 1H), 6.95 (dd, 1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99 (m, 1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96 (m, 1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30 (s, 3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73 (m, 1H), 1.46 (d, 3H). MS: m/z 617 [MH]+, as free base.
syn of intermediates

a(a) (i) 2-Bromo-5-fluorotoluene, Mg, THF, 60−70 °C; (ii) 4-methoxypyridine, benzyl chloroformate, THF, −20 °C, then Grignard’s reagent, −20 °C, 1 h; (b) (i) tris(triphenylphosphine)rhodium(I) chloride, 2-propanol, H2 (p = 5 atm), 60 °C, 5 h; (ii) Pd/C 5%, H2 (p = 4 atm), 20 °C, 5 h; (iii) (R,S)-10-camphorsulfonic acid, toluene; (c) CH2Cl2, H2O, 8% NaHCO3 (aq); l-(+)-mandelic acid, 2-propanol, heptanes; (d) MeNH2, EtOH, NaBH4, 25 °C, 1.5 h; (e) (i) ethyl acetate, NaHCO3 (aq. sat. soln), 5; (ii) triphosgene, triethylamine, ethyl acetate, then 5, 20 °C, 2 h; (f) R′RNH, CH3CN, NaBH(OAc)3, room temp, 24 h.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family