

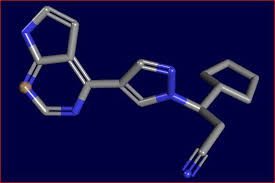
Ruxolitinib
(3R)-3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile, cas no 941678-49-5
(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile
Formula: C17H18N6
Molecular Weight: 306.37

Phosphate salt
(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate
INCB 018424
- INCB 018424
- INCB018424
- Ruxolitinib
- UNII-82S8X8XX8H
CAS No.: 1092939-17-7
M.Wt: 404.36
Formula: C17H21N6O4P
Ruxolitinib phosphate
LAUNCHED 2011, INCYTE FOR MYELOFIBROSIS, NDA202192, 2011-11-16
CLINICAL TRIALS.http://clinicaltrials.gov/search/intervention=INCB018424+OR+ruxolitinib
EMA:Link,
US FDA:link
HPLC, MS, NMR…http://www.medkoo.com/Product-Data/Ruxolitinib/Ruxolitinib-QC-LC20130225.pdf
http://file.selleckchem.com/downloads/nmr/S137803-INCB018424-HNMR-Selleck.pdf
http://file.selleckchem.com/downloads/hplc/S137803-INCB018424-HPLC-Selleck.pdf
Ruxolitinib phosphate is a kinase inhibitor with the chemical name (R)-3-(4-(7H-pyrrolo[2,3d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate and a molecular weight of 404.36.
Ruxolitinib is a janus-associated kinase inhibitor indicated to treat bone marrow cancer, specifically intermediate or high-risk myelofibrosis. FDA approved on November 16, 2011.
INCB018424 is the first potent, selective, JAK1/2 inhibitor to enter the clinic with IC50 of 3.3 nM/2.8 nM, >130-fold selectivity for JAK1/2 versus JAK3
Ruxolitinib phosphate has the following structural formula:
Ruxolitinib phosphate is a white to off-white to light pink powder and is soluble in aqueous buffers across a pH range of 1 to 8.
Jakafi (ruxolitinib) Tablets are for oral administration. Each tablet contains ruxolitinib phosphate equivalent to 5 mg, 10 mg, 15 mg, 20 mg and 25 mg of ruxolitinib free base together with microcrystalline cellulose, lactose monohydrate, magnesium stearate, colloidal silicon dioxide, sodium starch glycolate, povidone and hydroxypropyl cellulose
.NCI: /Ruxolitinib phosphate/ The phosphate salt form of ruxolitinib, an orally bioavailable Janus-associated kinase (JAK) inhibitor with potential antineoplastic and immunomodulating activities. Ruxolitinib specifically binds to and inhibits protein tyrosine kinases JAK 1 and 2, which may lead to a reduction in inflammation and an inhibition of cellular proliferation. The JAK-STAT (signal transducer and activator of transcription) pathway plays a key role in the signaling of many cytokines and growth factors and is involved in cellular proliferation, growth, hematopoiesis, and the immune response; JAK kinases may be upregulated in inflammatory diseases, myeloproliferative disorders, and various malignancies. (NCI Thesaurus)
patent expiry
US pat 7598257 exp 24/12/27
US pat 8415362 exp 24/12/27
NCE.Nov 16, 2016
Discovered by Incyte, ruxolitinib phosphate is an inhibitor of Janus-associated kinase 2 (JAK2), a protein involved in signal transduction. This orally available compound was approved and launched in the U.S. in 2011 for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MP and post-essential thrombocythemia MF. A regulatory application in the E.U. was filed in 2011 and a positive opinion was assigned in April 2012. Final E.U. approval was obtained in August 2012. In November 2012, the product was launched in the U.K. for the treatment of disease-related splenomegaly or symptoms in primary myelofibrosis or myelofibrosis due to polycythemia vera or essential thrombocythemia. In 2012, the product has been filed for approval in Japan for the treatment of myelofibrosis.
Phase II clinical trials are also ongoing for the treatment of multiple myeloma, leukemia, pancreas cancer, thrombocytopenia and for the treatment of relapsed or refractory diffuse large B-cell or peripheral T-cell non-Hodgkin lymphoma following donor stem cell transplant. In Japan, the product is under development in phase III trials for the treatment of polycythemia vera and in phase II trials for the treatment of myelofibrosis. No recent development has been reported for clinical trials for the treatment of rheumatoid arthritis (RA), for the treatment of psoriasis or for the treatment of metastatic prostate cancer. Columbia University is evaluating the compound in preclinical studies for the treatment of alopecia areata. The University of Pennsylvania is conducting phase II clinical trials for the treatment of breast cancer.
In 2008 and 2009, the compound was assigned orphan drug designation in the U.S. and the E.U., respectively, for the treatment of myelofibrosis. This designation was assigned in Japan for this indication in 2011. Additional orphan drug designation was assigned by the FDA in 2010 for the treatment of polycythemia vera and for the treatment of essential thrombocythemia. In 2013, an orphan drug designation was assigned in U.S. for the treatment of pancreatic cancer. In 2009, fast track designation was assigned to ruxolitinib phosphate in the U.S .for the treatment of myelofibrosis.
Ruxolitinib (trade names Jakafi and Jakavi, by Incyte Pharmaceuticals and Novartis) is a drug for the treatment of intermediate or high-risk myelofibrosis, a type of bone marrow cancer.It is also being investigated for the treatment of other types of cancer (such as lymphomas and pancreatic cancer), for polycythemia vera, and for plaque psoriasis.
The phase III Controlled Myelofibrosis Study with Oral JAK Inhibitor-I (COMFORT-I) and COMFORT-II trials showed significant benefits by reducing spleen size, relieving debilitating symptoms, and improving overall survival.
INCYTE developed in cooperation with companies and NOVARTIS jak2 selective inhibitor Ruxolitinib(INCB-018424) – has been approved by the FDA successfully listed). (Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. Srdan Verstovsek, MD, Ph.D., Hagop Kantarjian, MD, Ruben A. Mesa. MD, et al. N Engl J Med 2010; 363: 1117-1127).
The presently disclosed a series of patent applications JAK inhibitors, including WO2001042246, WO200200066K WO2009054941 and WO2011013785, etc.

Mechanism of action
Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes 1 and 2 of this enzyme.
Side effects
Immunologic side effects have included herpes zoster (1.9%) and case reports of opportunistic infections.[10] Metabolic side effects have included weight gain (7.1%). Laboratory abnormalities have included alanine transaminase (ALT) abnormalities (25.2%), aspartate transaminase (AST) abnormalities (17.4%), and elevated cholesterol levels (16.8%).
Legal status
In November 2011, ruxolitinib was approved by the USFDA for the treatment of intermediate or high-risk myelofibrosis based on results of the COMFORT-I and COMFORT-II Trials.
Some analysts believe this to be a potential blockbuster drug.[3] As of the end of March 2012, and according to an Incyte spokesman, approximately 1000 physicians had prescribed the drug in the United States, out of a total 6500 hematologists and oncologists nationwide.


The US Food and Drug Administration had approved Incyte’s Jakafi (ruxolitinib) to treat patients with the bone marrow disease myelofibrosis (MF). Jakafi is the first and only drug granted license specifically for the treatment of the rare blood cancer.
Jakafi approved by FDA to treat rare bone marrow disease
Posted By Edward Su On November 17th, 2011
MF is a rare, potentially life-threatening blood cancer with limited treatment methods. Patients with the bone marrow disoder, characterized by bone marrow failure, enlarged spleen (splenomegaly), suffer from the symptoms of fatigue, night sweats and pruritus, poor quality of life, weight loss and shortened survival. The US drug firm Incyte estimates the disease affects about 16,000-18,500 people in the USA. Currently, the disease is treated with chemotherapy or bone marrow transplant.
Incyte’s Jakafi, the first drug to reach market from the Wilmington-based drug company, was approved by the FDA as a twice-a-day pill for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF. The US regulators reviewed Jakafi under its priority review program for important new therapies.
The approval of Jakafi was based on the results from two clinical studies involved 528 patients with the disease. Patients in the Jakafi treatment arm experienced a significant reduction in the size of their spleen as well as a 50 percent decrease in symptoms, including pain, discomfort and night sweats.
Jakafi, generically known as ruxolitinib, works by blocking JAK1 and JAK2 enzymes associated with the disease. The company has co-developed the drug with Novartis as part of their collaboration signed in 2009. The Swiss drug firm has the rights to market Jakafi in other countries.
“The availability of Jakafi is a significant medical advancement for people living with myelofibrosis, a debilitating disease,” said Paul A. Friedman, M.D., President and Chief Executive Officer of Incyte. “This milestone marks a tremendous achievement for Incyte because a scientific discovery from our research laboratories has become the first JAK inhibitor to reach the market and provide a clinical benefit to patients.”
Richard Pazdur, director of the Office of Hematology and Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said that Jakafi “represents another example of an increasing trend in oncology where a detailed scientific understanding of the mechanisms of a disease allows a drug to be directed toward specific molecular pathways”.
Incyte says Jakafi will be available next week, and the drug will cost $7,000 per month, or $84,000 for a year’s supply for insured patients. The company plans to provide Jakafi free to uninsured patients and will offer co-pay assistance to patients with financial need.
……………
NMR free base
http://www.google.com/patents/US8410265
For (R)-13 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6 (MW, 306.37) LCMS (EI) m/e 307 (M++H).
http://www.google.com/patents/US8410265
phosphate
For (R)-14 (phosphate): mp. 197.6° C.; 1H NMR (DMSO-d6, 500 MHz) δ ppm 12.10 (s, 1H), 8.78 (s, 1H), 8.68 (s, 1H), 8.36 (s 1H), 7.58 (dd, 1H, J=1.9, 3.5 Hz), 6.97 (d, 1H, J=3.6 Hz), 4.52 (td, 1H, J=3.9, 9.7 Hz), 3.25 (dd, 1H, J=9.8, 17.2 Hz), 3.16 (dd, 1H, J=4.0, 17.0 Hz), 2.41, (m, 1H), 1.79 (m, 1H), 1.59 (m, 1H), 1.51 (m, 2H), 1.42 (m, 1H), 1.29 (m, 2H), 1.18 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ ppm 152.1, 150.8, 149.8, 139.2, 131.0, 126.8, 120.4, 118.1, 112.8, 99.8, 62.5, 44.3, 29.1, 29.0, 24.9, 24.3, 22.5; C17H18N6(MW, 306.37 for free base) LCMS (EI) m/e 307 (M++H, base peak), 329.1 (M++Na).
………………….
SYNTHESIS
http://www.google.com/patents/US8410265



………………………
SYNTHESIS
US20100190981
(R)-Methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilypethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoate ((R)-22). A solution of (E)-methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)acrylate (21, 815 mg) in tetrahydrofuran (THF, 8.0 mL) in a pressure glass tube was treated with the catalyst [Rh(COD)(−)-DuanPhos](BF4) (4.6 mg) under nitrogen before the reaction mixture was pressurized with hydrogen gas to 50 bar pressure. The reaction mixture was stirred at 35° C. under this hydrogen pressure for 22 h. When HPLC analysis showed that the substrate was almost completely consumed, the reaction mixture was cooled down to room temperature. The enantiomeric excess of the reaction mixture was determined to be 94.7% ee (97.35% of the second peak, (R)-22; 2.65% of the first peak, (S)-22) by chiral HPLC analysis. The reaction mixture was then filtered through a thin silica gel pad and the pad was washed with tetrahydrofuran (THF, 5 mL). The filtrate was then concentrated under reduced pressure to dryness. The resultant foamy solid (778 mg) was analyzed by chiral HPLC analysis and result showed a 94.7% of enantiomeric excess favoring the second peak (97.35% of the second peak, (R)-22; 2.65% of the first peak, (S)-22).

(3R)-3-Cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23). To a stirred solution of (3R)-methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoate ((R)-22, 2.47 g, 5.26 mmol) in THF (30 mL) at room temperature was added a solution of lithium hydroxide monohydrate (LiOH—H2O, 265 mg, 6.31 mmol, 1.2 equiv) in water (15 mL). The reaction mixture was stirred at room temperature for 3 h. When LCMS showed the reaction was complete, the reaction mixture was then acidified with 1 N aqueous HCl solution to pH 5 before it was extracted with EtOAc (2×25 mL). The combined organic layers were washed with brine, dried over magnesium sulfate (MgSO4), filtered and concentrated under reduced pressure to afford (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23, 2.40 g, 2.40 g theoretical, 100% yield) as a colorless oil, which solidified upon standing at room temperature in vacuo. For (R)-23: 1H NMR (CDCl3, 300 MHz) δ ppm 8.95 (s, 1H), 8.95 (bs, 1H), 8.36 (s, 1H), 7.57 (d, 1H, J=3.7 Hz), 6.99 (d, 1H, J=3.7 Hz), 5.74 (s, 2H), 4.65 (dt, 1H, J=3.1, 10.3 Hz), 3.58 (t, 2H, J=8.2 Hz), 3.24 (dd, 1H, J=16.5, 10.3 Hz), 3.04 (dd, 1H, J=16.2, 3.1 Hz), 2.59 (m, 1H), 2.00 (m, 1H), 1.77-1.24 (m, 7H), 0.97 (t, 2H, J=8.2 Hz), 0.00 (s, 9H); C23H33N5O3Si (MW, 455.63), LCMS (EI) m/e 456.1 (M++H).
(3R)-3-Cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24). To a stirred solution of (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23, 20 mg, 0.044 mmol) in DMF (1 mL) at room temperature was added N,N-carbonyldiimidazole (CDI, 21 mg, 0.13 mmol, 3.0 equiv). The reaction mixture was then stirred at room temperature and TLC was used to follow the reaction for formation of acyl imidazole (consumption of acid to a higher Rf spot with 30% EtOAc/hexane). When TLC showed that the acyl imidazole transformation was complete, ammonia gas was then bubbled through the stirred solution for 30 min to afford the amide (followed by LCMS). The excess amount of ammonia gas was evaporated by bubbling nitrogen vigorously through the solution. The crude product, (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24), in DMF was used directly to the following reaction to convert amide ((R)-24) into the corresponding nitrile ((R)-20).
(3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20). Method A. To a stirred solution of (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24, 20 mg, 0.044 mmol) in DMF (1 mL) at 0° C. was added methylene chloride (1 mL) and triethylamine (0.12 mL, 0.88 mmol, 20.0 equiv), followed by trichloroacetyl chloride (0.052 ml, 0.462 mmol, 10.5 equiv). The resulting reaction mixture was stirred at 0° C. for 1 h. When LCMS showed the reaction was complete, the reaction mixture was quenched with saturated sodium bicarbonate solution (NaHCO3, 5 mL) before being extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine, dried over magnesium sulfate (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography with 0-75% EtOAc/hexane gradient elution to give (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20, 10 mg, 19 mg theoretical, 53% yield). For (R)-20: 1H NMR (DMSO-d6, 400 MHz) δ ppm 8.83 (s, 1H), 8.75 (s, 1H), 8.39 (s, 1H), 7.77 (d, 1H, J=3.7 Hz), 7.09 (d, 1H, J=3.7 Hz), 5.63 (s, 2H), 4.53 (td, 1H, J=19.4, 4.0 Hz), 3.51 (t, 2H, J=8.1 Hz), 3.23 (dq, 2H, J=9.3, 4.3 Hz), 2.41 (m, 1H), 1.79 (m, 1H), 1.66-1.13 (m, 7H), 0.81 (t, 2H, J=8.2 Hz), 0.124 (s, 9H); C23H32N6OSi (MW, 436.63), LCMS (EI) m/e 437 (M−+H) and 459 (M++Na).
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-13, free base). Method B. To a solution of (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L) was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4, 987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The reaction temperature was observed to decrease from ambient to 12° C. upon addition of the water and then increase to 33° C. during the addition of lithium tetrafluoroborate (LiBF4). The resulting reaction mixture was heated to reflux (about 80° C.) for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC analyses showed both the hydroxyl methyl intermediate ((R)-25) and fully de-protected material ((R)-13, free base) produced but no starting material ((R)-20) left, the reaction mixture was cooled gradually to <5° C. before a 20% aqueous solution of ammonium hydroxide (NH4OH, 450 mL) was added gradually to adjust the pH of the reaction mixture to 9 (checked with pH strips). The cold bath was removed and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v) to confirm complete de-protection. When LCMS and TLC showed the reaction was deemed complete, the reaction mixture was filtered and the solids were washed with acetonitrile (1 L). The combined filtrates were then concentrated under reduce pressure, and the residue was partitioned between ethyl acetate (EtOAc, 6 L) and half-saturated brine (3 L). The two layers were separated and the aqueous layer was extracted with ethyl acetate (2 L). The combined organic layers were washed with half-saturated sodium bicarbonate (NaHCO3, 3 L) and brine (3 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure to give the crude product as an orange oil. The crude material was then purified by flash column chromatography (SiO2, 40 to 100% ethyl acetate/heptane gradient elution) to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-13, free base, 273 g, 324.9 g theoretical, 84% yield) as a white foam. This material was checked by 19F NMR to ensure no lithium tetrafluoroborate (LiBF4) remained and by chiral HPLC (Chiralcel OD, 90:10 hexane/ethanol) to confirm enantiomeric purity and was used without further purification to prepare the corresponding phosphate salt. For (R)-13 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6(MW, 306.37) LCMS (EI) m/e 307 (M++H).

3-Cyclopentylacrylonitrile (8). A solution of diethyl cyanomethylphosphonate (7, 742.5 g, 4.2 mol, 1.1 equiv) in dry THF (5.75 L) was stirred under nitrogen on an ice-water-methanol bath and a solution of 1 M potassium tert-butoxide in THF (4 L, 4.0 mol, 1.05 equiv) was added at such a rate as to keep the temperature below 0° C. After addition of 1 M potassium tert-butoxide in THF was complete, the stirring was continued on the cold bath for 1 h and a solution of cyclopentanecarbaldehyde (6, 374 g, 3.81 mol) in dry THF (290 mL) was added at such a rate as to maintain the temperature below 0° C. The cold bath was removed, and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. When the reaction was deemed complete, the reaction mixture was partitioned between methyl tent-butyl ether (MTBE, 14 L), water (10 L) and brine (6 L). The two layers were separated, and the combined organic phase was washed with brine (6 L). The aqueous phase was extracted with MTBE (10 L) and washed with brine (6 L). The combined organic extracts were concentrated under reduced pressure and the residue was distilled (65-78° C./6 torr) to afford 3-cyclopentylacrylonitrile (8, 437.8 g, 461.7 g theoretical, 94.8% yield) as a colorless oil, which was found to be a mixture of E- and Z-isomer. For 8: 1H NMR (DMSO-d6, 400 MHz, for Z-isomer) δ ppm 6.58 (t, 1H, J=10.6 Hz), 5.55 (dd, 1H, J=10.8, 0.59 Hz), 2.85 (m, 1H), 1.9-1.46 (m, 6H), 1.34 (m, 2H) and (for E-isomer) δ ppm 6.83 (q, 1H, J=8.3 Hz), 5.66 (dd, 1H, J=16.5, 1.4 Hz), 2.60 (m, 1H), 1.9-1.46 (m, 6H), 1.34 (m, 2H); 13C NMR (DMSO-d6, 100 MHz, for Z-isomer) δ ppm 159.8, 116.6, 97.7, 42.3, 32.3, 25.1 and (for E-isomer) δ ppm 160.4, 118.1, 97.9, 43.2, 31.5, 24.8; C8H11N (MW, 121.18), GCMS (EI) m/e 120 (M+−H).


4-(1H-Pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5). Method A. To a flask equipped with a reflux condenser, a nitrogen inlet, mechanical stirrer, and a thermowell was added 4-chloro-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (3a, 817 g, 2.88 mol) and dioxane (8 L). To this solution was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 728 g, 3.75 mol, 1.30 equiv) followed by a solution of potassium carbonate (K2CO3, 1196 g, 8.67 mol, 3.0 equiv) in water (4 L). The solution was degassed by passing a stream of nitrogen through the solution for 15 minutes before being treated with tetrakis(triphenylphosphine)palladium(0) (167 g, 0.145 mol, 0.05 equiv) and the resulting reaction mixture was heated at reflux (about 90° C.) for 2 hours. When the reaction was deemed complete by TLC (1:1 heptane/ethyl acetate) and LCMS, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (24 L) and water (4 L). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (4 L). The combined organic layers were washed with water (2×2 L), brine (2 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure. The residue was suspended in toluene (4 L) and the solvent was removed under reduced pressure. The residue was finally triturated with methyl tert-butyl ether (MTBE, 3 L) and the solids were collected by filtration and washed with MTBE (1 L) to afford 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanyl-ethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 581.4 g, 908.5 g theoretical, 64% yield) as white crystalline solids. For 5: 1H NMR (DMSO-d6, 400 MHz) δ ppm 13.41 (bs, 1H), 8.74 (s, 1H), 8.67 (bs, 1H), 8.35 (bs, 1H), 7.72 (d, 1H, J=3.7 Hz), 7.10 (d, 1H, J=3.7 Hz), 5.61 (s, 2H), 3.51 (t, 2H, J=8.2 Hz), 0.81 (t, 2H, J=8.2 Hz), 0.13 (s, 9H); C15H21N5OSi (MW, 315.45), LCMS (EI) m/e 316 (M++H).
Racemic 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound). Method A. 3-Cyclopentylacrylonitrile (8, 273.5 g, 2.257 mol, 1.20 equiv) and DBU (28 mL, 0.187 mol, 0.10 equiv) was added to a suspension of 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 591.8 g, 1.876 mol) in acetonitrile (4.7 L) at room temperature. The resulting reaction mixture was heated to 50-60° C. for 17 hours (a clear solution developed midway through heating) then to 70-80° C. for 8 hours. When LCMS analysis showed the reaction was deemed complete, the reaction mixture was cooled to room temperature. The cooled solution was then concentrated under reduced pressure to give the crude product (9) as a thick amber oil. The crude product was dissolved in dichloromethane (DCM) and absorbed onto silica gel then dry-loaded onto a silica column (3 Kg) packed in 33% EtOAc/heptanes. The column was eluted with 33% EtOAc/heptanes (21 L), 50% EtOAc/heptanes (28 L), 60% EtOAc/heptanes (12 L) and 75% EtOAc/heptanes (8 L). The fractions containing the desired product (9) were combined and concentrated under reduced pressure to generate a yellow oil, which was transferred to a 3 L flask with EtOAc. The solvent was removed under reduced pressure and the residual EtOAc by co-evaporating with heptanes. The residue was further dried under high vacuum for overnight to afford racemic 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound, 800 g, 819.1 g theoretical, 97.7% yield) as an extremely viscous yellow oil. For 9: 1H NMR (DMSO-d6, 400 MHz) δ ppm 8.83 (s, 1H), 8.75 (s, 1H), 8.39 (s, 1H), 7.77 (d, 1H, J=3.7 Hz), 7.09 (d, 1H, J=3.7 Hz), 5.63 (s, 2H), 4.53 (td, 1H, J=19.4, 4.0 Hz), 3.51 (t, 2H, J=8.1 Hz), 3.23 (dq, 2H, J=9.3, 4.3 Hz), 2.41 (m, 1H), 1.79 (m, 1H), 1.66-1.13 (m, 7H), 0.81 (t, 2H, J=8.2 Hz), 0.124 (s, 9H); C23H32N6OSi (MW, 436.63), LCMS (EI) m/e 437 (M++H) and 459 (M++Na).
(3R)-Cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo [2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10) and (3S)-Cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((S)-10) A slurry of 1.5 Kg of 20-micron Chiralcel® OD chiral stationary phase (CSP) made by Daicel in 3.0 L of isopropanol (IPA) was packed into a PROCHROM Dynamic Axial Compression Column LC110-1 (11 cm ID×25 cm L; Column Void Vol.: approximate 1.5 L) under 150 bar of packing pressure. The packed column was then installed on a Novasep Hipersep HPLC unit. The column and the Hipersep unit were flushed with methanol (17 L) followed by the mobile phase made of a mixture of isopropanol and hexane (2:8 by volume, 17 L). The feed solution was then prepared by dissolving 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound, 2795 g, 6.4 mol) in the mobile phase to a concentration of 80 g/L. The feed solution was then sequentially injected into the preparative chiral column for separation. Each injection was 120 ml in volume. The chiral column was eluted with the mobile phase at a flow rate of 570 mL/min at room temperature. The column elution was monitored by UV at a wavelength of 330 nm. Under these conditions a baseline separation of the two enantiomers was achieved. The retention times were 16.4 minutes (Peak 1, the undesired (S)-enantiomer (S)-10) and 21.0 minutes (Peak 2, the desired (R)-enantiomer (R)-10), respectively. The cycle time for each injection was 11 minutes and a total of 317 injections were performed for this separation process. Fractions for Peak 1 (the undesired (S)-enantiomer, (S)-10) and Peak 2 (the desired (R)-enantiomer, (R)-10) were collected separately from each injection. The collected fractions collected were continuously concentrated in the 1-square feet and 2-square feet ROTOTHERM evaporator, respectively, at 40° C. under reduced pressure (40-120 bar). The residue from each evaporator was further dried under high vacuum to constant weight to afford (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10, 1307 g, 1397.5 g theoretical, 93.5%) from Peak 2 as a light yellow oil and (3S)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((S)-10, 1418 g, 1397.5 g theoretical, 101.5%) from Peak 1 as an yellow oil.
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base). Method A. To a solution of (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L) was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4, 987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The reaction temperature was observed to decrease from ambient to 12° C. upon addition of the water and then increase to 33° C. during the addition of lithium tetrafluoroborate (LiBF4). The resulting reaction mixture was heated to reflux (about 80° C.) for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC analyses showed both the hydroxyl methyl intermediate ((R)-11) and fully de-protected material ((R)-12, free base) produced but no starting material ((R)-10) left, the reaction mixture was cooled gradually to <5° C. before a 20% aqueous solution of ammonium hydroxide (NH4OH, 450 mL) was added gradually to adjust the pH of the reaction mixture to 9 (checked with pH strips). The cold bath was removed and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v) to confirm complete de-protection. When LCMS and TLC showed the reaction was deemed complete, the reaction mixture was filtered and the solids were washed with acetonitrile (1 L). The combined filtrates were then concentrated under reduce pressure, and the residue was partitioned between ethyl acetate (6 L) and half-saturated brine (3 L). The two layers were separated and the aqueous layer was extracted with ethyl acetate (2 L). The combined organic layers were washed with half-saturated sodium bicarbonate (NaHCO3, 3 L) and brine (3 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure to give the crude product as an orange oil. The crude material was then purified by flash column chromatography (SiO2, 40 to 100% ethyl acetate/heptane gradient elution) to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base, 273 g, 324.9 g theoretical, 84% yield) as a white foam. This material was checked by 19F NMR to ensure no lithium tetrafluoroborate (LiBF4) remained, and by chiral HPLC (Chiralcel® OD-H, 90:10 hexane/ethanol) to confirm enantiomeric purity (98.7% ee), and was used without further purification to prepare the corresponding phosphate salt. For (R)-12 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6(MW, 306.37) LCMS (EI) m/e 307 (M++H).

(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile (R)-10. A solution of (R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanenitrile ((R)-10, 75.0 g, 0.172 mol, 98.8% ee) in acetonitrile (600 mL) was cooled to 0-5° C. To the cooled solution was added boron trifluoride diethyl etherate (54.4 mL, 0.429 mol) over 10 minutes while maintaining the internal reaction temperature below 5° C. Following the addition, the cold bath was removed and the reaction mixture was allowed to warm to room temperature. When HPLC analysis indicated that the level of (R)-10 was below 1%, the initial phase of the deprotection reaction was considered complete. The reaction was then cooled to 0-5° C., followed by the slow addition of water (155 mL). Following the water addition, the cold bath was removed and the resulting reaction mixture was allowed to warm to 13-17° C., and stirred for an additional 2-3 hours. The resulting reaction mixture was cooled again to 0-5° C. To the cooled reaction mixture was added slowly a solution of ammonia in water [prepared by mixing aqueous 28% ammonia solution (104.5 mL) and water (210.5 mL)] while maintaining the internal reaction temperature at below 5° C. After the aqueous ammonia solution was added, the cold bath was removed and the reaction was allowed to warm to room temperature. The hydrolysis was deemed complete when the level of the hydroxylmethyl intermediate was below 1% by HPLC analysis.
The resulting reaction mixture was diluted with ethyl acetate (315 mL) and washed with 20% brine (315 mL). The aqueous fraction was back extracted with ethyl acetate (315 mL). The organic fractions were combined and concentrated under vacuum with a bath temperature of 40° C. to a volume of 380 mL. The concentrated residue was diluted with ethyl acetate (600 mL) and washed with 1M NaHCO3 (2×345 mL) and 20% brine (345 mL). The aqueous washes were combined and back extracted with ethyl acetate (345 mL). The organic fractions were combined and polish filtered into a clean 2L round bottom flask. The organic fraction was washed with warm water (50° C., 2×450 mL) and then treated with activated charcoal at 65° C. with stirring for 1.5 hours. The slurry was filtered through a celite bed. The filtrate was concentrated under vacuum with a bath temperature of 40° C. The resulting syrup was placed under high vacuum to provide (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile [(R)-12, 54.2g, 103% yield] as a light yellow foam. This material was checked by 19F NMR to ensure that the product was not contaminated by any fluorinated impurities. The chemical purity of the isolated free base was 96.3%. The chiral purity of the free base was 98.8% by chiral HPLC (chiralcel OD, 90:10 hexane/ethanol). The free base was used without further purification to prepare the phosphate salt. 1H NMR (DMSO-d6, 400 MHz) δ 12.11(bs, 1H), 8.79(d, 1H, J=0.43 Hz), 8.67(s, 1H), 8.37(s, 1H), 7.59(q, 1H, J=2.3 Hz), 6.98(q, 1H, J=1.6 Hz), 4.53(td, 1H, J=19.2, 4.1 Hz), 3.22(dq, 2H, J=9.8, 4.3 Hz), 2.40(m, 1H), 1.79(m, 1H), 1.65-1.13(m, 7H). C17H16N6 (MW, 306.37), LCMS (EI) m/e 307 (M++H).

(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile phosphate salt ((R)-13, phosphate).
Method A. To a solution of (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base, 572 g, 1.87 mol) in isopropanol (IPA, 8 L) at 60-65° C. was added a solution of phosphoric acid (186.2 g, 1.9 mol, 1.10 equiv) in isopropanol (1.6 L). No exotherm was observed while adding a solution of phosphoric acid, and a precipitate was formed almost immediately. The resulting mixture was then heated at 76° C. for 1.5 hours, then cooled gradually to ambient temperature and stirred at room temperature for overnight. The mixture was filtered and the solids were washed with a mixture of heptanes and isopropanol (1/1, v/v, 3 L) before being transferred back to the original flask and stirred in heptanes (8 L) for one hour. The solids were collected by filtration, washed with heptanes (1 L), and dried in a convection oven in vacuum at 40° C. to a constant weight to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile phosphate salt ((R)-13, phosphate, 634.2 g , 755 g theoretical, 84% yield) as white to off-white crystalline solids. For (R)-13, phosphate: mp. 197.6° C.; 1H NMR (DMSO-d6, 500 MHz) δ ppm 12.10 (s, 1H), 8.78 (s, 1H), 8.68 (s, 1H), 8.36 (s 1H), 7.58 (dd, 1H, J=1.9, 3.5 Hz), 6.97 (d, 1H, J=3.6 Hz), 4.52 (td, 1H, J=3.9, 9.7 Hz), 3.25 (dd, 1H, J=9.8, 17.2 Hz), 3.16 (dd, 1H, J=4.0, 17.0 Hz), 2.41, (m, 1H), 1.79 (m, 1H), 1.59 (m, 1H), 1.51 (m, 2H), 1.42 (m, 1H), 1.29 (m, 2H), 1.18 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ ppm 152.1, 150.8, 149.8, 139.2, 131.0, 126.8, 120.4, 118.1, 112.8, 99.8, 62.5, 44.3, 29.1, 29.0, 24.9, 24.3, 22.5; C17H18N6(MW, 306.37 for free base) LCMS (EI) m/e 307 (M++H, base peak), 329.1 (M++Na).
Method B. To a solution of (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H -pyrazol-1-yl)-3-cyclopentylpropanenitrile ((R)-12, 54.2 g, 177 mol) in dichloromethane (782 mL) and 2-propanol (104 mL) at reflux was added a solution of phosphoric acid (19.9 g, 0.173 mol, 1.15 equiv) in 2-propanol (34.0 mL) over a period of 47 minutes. Following the acid addition, the resulting mixture was heated to reflux for an additional 1 hour. The mixture was gradually cooled to ambient temperature and stirred for 3 hours. The solids were collected by filtration and washed with dichloromethane (390 mL), followed by n-heptane (390 mL). The solids were partially dried under vacuum at room temperature and then under vacuum at 62° C. to afford (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate (60.1 g, 84% yield) as white to off-white crystalline solids. Analysis by chiral HPLC (chiralcel OD, 90:10 hexane/ethanol) gave the enantiopurity as 99.2% ee.
1H NMR (DMSO-d6, 400 MHz) δ 12.11(bs, 1H), 8.79(d, 1H, J=0.59 Hz), 8.67(s, 1H), 8.36(s, 1H), 7.59(q, 1H, J=2.3 Hz), 6.98(q, 1H, J=1.6 Hz), 4.53(td, 1H, J=19.6, 4.4 Hz), 3.22(dq, 2H, J=9.6, 4.3 Hz), 2.40(m, 1H), 1.79(m, 1H), 1.65-1.13(m, 7H). C17H21N6O4P (MW, 404.36), LCMS (EI) m/e 307 (M++H) and m/e 329 (M++Na).
(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate.
Into a 1L round bottom flask, equipped with stir bar, distillation head, addition funnel and heating mantle, were charged methanol (520 mL) and (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate ((R)-13, phosphate, 40.0 grams, 98.92 mmol). The slurry was heated to 55° C. to generate a slightly pink solution. The solution was cooled to 50° C. and filtered into a 2 L flask equipped with an overhead stirrer, distillation head, addition funnel and heating mantle. The 1 L round bottom flask and the filter funnel were rinsed with additional methanol (104.0 mL). The filtrate solution was heated to reflux to distill methanol (281 mL) over 1 hour under atmospheric pressure. Isopropyl alcohol (IPA) (320 mL) was charged slowly via the addition funnel over 80 minutes while maintaining the internal temperature approximately at 65° C. Precipitation of the phosphate salt was observed during IPA addition. After the addition of IPA was complete, n-heptane (175 mL) was added slowly at the same temperature. Distillation was continued under atmospheric pressure. Additional n-heptane (825 mL) was added at approximately the same rate as the distillation rate while maintaining the internal temperature at approximately 65° C. The distillation was complete when the volume of the distillate reached 742 mL (excluding the volume of 281 mL of methanol from the previous distillation). The distillation took approximately 1 hour. The vapor temperature during the distillation was in the range of 54-64° C. and the internal temperature was 67° C. at the end of the distillation. The mixture was slowly cooled to room temperature and stirred for an additional 3 hours. The solids were collected by filtration. The wet cake was washed with 16.7% (v/v) of isopropyl alcohol in n-heptane (384.0 mL), followed by n-heptane (280.0 mL), and dried under vacuum at 55° C. to provide 36.1 grams of the desired product as white solids in 90% yield. The chemical purity is 99.79% by HPLC analysis. The chiral purity is 99.8% by chiral HPLC analysis.
1H NMR (499.7 MHz, DMSO-d6) δ (ppm): 12.21 (s, 1H), 10.71 (s, 3H), 8.80 (s, 1H), 8.72 (s, 1H), 8.40 (s, 1H), 7.60 (d, J=3.5 Hz, 1H), 7.00 (d, J=3.5 Hz, 1H), 4.51 (td, J=9.75, 4.0 Hz, 1H), 3.25 (dd, J=17.3, 9.75 Hz, 1H), 3.14 (dd, J=17.0, 4.0 Hz, 1H), 2.43-2.35 (m, 1H), 1.79-1.73 (m, 1H), 1.58-1.42 (m, 3H), 1.41-1.33 (m, 1H), 1.30-1.23 (m, 2H), 1.19-1.12 (m, 1H);
13C NMR (125.7 MHz, DMSO-d6) δ (ppm): 152.8, 151.2, 150.3, 140.0, 131.8, 127.7, 120.8, 118.8, 113.5, 100.7, 63.3, 45.0, 29.8, 25.6, 25.0, 23.2;
LCMS m/z: calculated for C17H18N6 (M+H)+:=307.2. Found (M+H)+: 307.0.
……………………….
US8410265
(JAK1, JAK2) inhibitor, developed by the Incyte Corporation, trade name Jakafi.
Ruxolitinib synthetic route as shown below. 4 – bromo-pyrazole ( 1 ) with ethyl vinyl ether ( 2 ) to protect, and then with a Grignard reagent to a halogen – exchanged with isopropyl magnesiumpinacol ester ( 3 ) quenching to obtain 4 . Compound 5 is obtained consisting of hydrogen is protected 6 , and then with a boronic acid ester 4 Suzuki coupling occurs under acidic conditions after removal of the protecting group pyrazolyl 7 , 7 and α, β-unsaturated aldehyde 8 chiral catalyst 9 of under the catalysis of asymmetric Michael addition to give ( R ) -10 (90% EE). ( R) -10 , after reaction with ammonia to obtain an imine oxidation with iodine nitrile 11 , respectively, with different conditions for the final removal of the protecting group to afford Ruxolitinib.

…………………………………
Bioorganic and Medicinal Chemistry Letters, 2013 , vol. 23, # 9 p. 2793 – 2800
http://www.sciencedirect.com/science/article/pii/S0960894X13001868

Figure 1.
Structures of tofacitinib (1a) and ruxolitinib (1b).
………………………….
Organic Letters, 2009 , vol. 11, 9 p. 1999 – 2002
http://pubs.acs.org/doi/abs/10.1021/ol900350k
An enantioselective synthesis of INCB018424 via organocatalytic asymmetric aza-Michael addition of pyrazoles (16 or 20) to (E)-3-cyclopentylacrylaldehyde (23) using diarylprolinol silyl ether as the catalyst was developed. Michael adducts (R)-24 and (R)-27 were isolated in good yield and high ee and were readily converted to INCB018424
http://pubs.acs.org/doi/suppl/10.1021/ol900350k/suppl_file/ol900350k_si_001.pdf
COMPD 1 IS RUXOLITINIB
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol
-1-yl]propionitrile (1, INCB018424).
. Method A. To a solution of (3R)-cyclopentyl-3-
{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-
yl}proprionitrile ((R)-25, INCB032306, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L)
was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4,
987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The resulting reaction mixture was
heated to reflux for overnight. An aliquot was quenched into ethyl acetate/water and
checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC
analyses indicated that both the hydroxyl methyl intermediate (R)-26, INCB021499) and
the fully deprotected product (1, INCB018424) SEE LINKhttp://pubs.acs.org/doi/suppl/10.1021/ol900350k/suppl_file/ol900350k_si_001.pdf
:1H NMR
(DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J = 0.4 Hz), 8.67 (s, 1H), 8.37
(s, 1H), 7.59 (dd, 1H, J = 2.3, 3.5 Hz), 6.98 (dd, 1H, J = 1.4, 3.4 Hz), 4.53 (td, 1H, J =
19.5, 4.6 Hz), 3.26 (dd, 1H, J = 9.8, 17.2 Hz), 3.18 (dd, 1H, J = 4.3, 17.3 Hz), 2.40 (m, S-12
1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H);
13C NMR (DMSO-d6, 100MHz) δ ppm 152.1,
151.0, 149.9, 139.3, 131.0, 126.8, 120.6, 118.2, 112.8, 99.8, 62.5, 44.3, 29.1, 25.0, 24.3,
22.5; IR (KBr) 3197, 3118, 2956, 2865, 1731, 1581, 1448, 1344, 1251 cm-1;
HRMS (CI)
m/z calculated for C17H19N6 (M + H)+
307.1671, found 307.1665
REFERENCES
- Jakafi (ruxolitinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 16 February 2014.
- Mesa, Ruben A.; Yasothan, Uma; Kirkpatrick, Peter (2012). “Ruxolitinib”. Nature Reviews Drug Discovery 11 (2): 103–4.doi:10.1038/nrd3652. PMID 22293561.
- Harrison, C; Mesa, R; Ross, D; Mead, A; Keohane, C; Gotlib, J; Verstovsek, S (2013). “Practical management of patients with myelofibrosis receiving ruxolitinib”. Expert Review of Hematology 6 (5): 511–23. doi:10.1586/17474086.2013.827413. PMID 24083419.
- Natoli, Cori Anne (May 5, 2012), “Incyte looks to ride on drug’s success”, The News Journal, retrieved May 6, 2012
- Harrison, C.; Kiladjian, J. J.; Al-Ali, H. K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D. S.; Levy, R.; Knoops, L.; Cervantes, F.; Vannucchi, A. M.; Barbui, T.; Barosi, G. (2012). “JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis”. New England Journal of Medicine 366 (9): 787–798.doi:10.1056/NEJMoa1110556. PMID 22375970. edit
- Verstovsek, S.; Mesa, R. A.; Gotlib, J.; Levy, R. S.; Gupta, V.; Dipersio, J. F.; Catalano, J. V.; Deininger, M.; Miller, C.; Silver, R. T.; Talpaz, M.; Winton, E. F.; Harvey Jr, J. H.; Arcasoy, M. O.; Hexner, E.; Lyons, R. M.; Paquette, R.; Raza, A.; Vaddi, K.; Erickson-Viitanen, S.; Koumenis, I. L.; Sun, W.; Sandor, V.; Kantarjian, H. M. (2012). “A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis”. New England Journal of Medicine 366 (9): 799–807. doi:10.1056/NEJMoa1110557.PMID 22375971. edit
- Tefferi, A. (2012). “Challenges Facing JAK Inhibitor Therapy for Myeloproliferative Neoplasms”. New England Journal of Medicine 366(9): 844–846. doi:10.1056/NEJMe1115119. PMID 22375977. edit
- ASCO Annual Meeting 2011: JAK Inhibitor Ruxolitinib Demonstrates Significant Clinical Benefit in Myelofibrosis
- Mesa, RA (2010). “Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis”. IDrugs : the investigational drugs journal 13 (6): 394–403.PMID 20506062. edit
- Pardanani, A.; Tefferi, A. (2011). “Targeting myeloproliferative neoplasms with JAK inhibitors”. Current Opinion in Hematology 18 (2): 1. doi:10.1097/MOH.0b013e3283439964. PMID 21245760. edit
- Wysham, NG; Allada G, Sullivan DR (2013). Chest 143 (5): 1478–9.PMID 23648912.
- “FDA Approves Incyte’s Jakafi(TM) (ruxolitinib) for Patients with Myelofibrosis” (Press release). Incyte. Retrieved 2012-01-02.
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H. K.; Gisslinger, H.; Waltzman, R.;Stalbovskaya, V.; McQuitty, M.; Hunter, D. S.; Levy, R.; Knoops, L.;Cervantes, F.; Vannucchi, A. M.; Barbui, T.; Barosi, G. N. Eng. J. Med. 2012,366, 787.Zhou, J.; Liu, P.; Lin, Q.; Metcalf, B. W.; Meloni, D.; Pan, Y.; Xia, M.; Li, M.; Yue,T.-Y.; Rodgers, J. D.; Wang, H. WO 2010083283 A2, 2010.Rodgers, J. D.; Shepard, S.; Maduskuie, T. P.; Wang, H.; Falahatpisheh, N.;Rafalski, M.; Arvanitis, A. G.; Storace, L.; Jalluri, R. K.; Fridman, J. S.; Vaddi, K.U.S. 20070135461 A1, 2007.Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.;Metcalf, B.; Yue, T.-Y.; Liu, P.; Zhou, J. Org. Lett. 1999, 2009, 11.http://www.google.com/patents/US8410265
| US7598257 * |
Dec 12, 2006 |
Oct 6, 2009 |
Incyte Corporation |
Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US20090233903 * |
Mar 10, 2009 |
Sep 17, 2009 |
Incyte Corporation |
Azetidine and cyclobutane derivatives as jak inhibitors |
| WO2007070514A1 * |
Dec 12, 2006 |
Jun 21, 2007 |
Incyte Corp |
Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| WO2007117494A1 * |
Apr 5, 2007 |
Oct 18, 2007 |
Vertex Pharma |
Deazapurines useful as inhibitors of janus kinases |
| US8309718 |
Nov 13, 2008 |
Nov 13, 2012 |
Incyte Corporation |
4-pyrazolyl-N-arylpyrimidin-2-amines and 4-pyrazolyl-N-heteroarylpyrimidin-2-amines as janus kinase inhibitors |
| US8410265 |
Jan 14, 2010 |
Apr 2, 2013 |
Incyte Corporation |
Processes for preparing JAK inhibitors and related intermediate compounds |
| US8415362 |
Jun 12, 2008 |
Apr 9, 2013 |
Incyte Corporation |
Pyrazolyl substituted pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8486902 |
Oct 8, 2010 |
Jul 16, 2013 |
Incyte Corporation |
Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8530485 |
Mar 30, 2011 |
Sep 10, 2013 |
Incyte Corporation |
Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8541425 |
Aug 27, 2009 |
Sep 24, 2013 |
Incyte Corporation |
Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8604043 |
May 21, 2010 |
Dec 10, 2013 |
Incyte Corporation |
3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane- or heptane-nitrile as jak inhibitors |
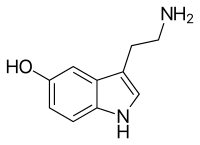




 The chemical name for serotonin is 5-hydoxytryptamine which is often abbreviated to 5-HT.
The chemical name for serotonin is 5-hydoxytryptamine which is often abbreviated to 5-HT.


 into the synaptic cleft. The serotonin molecules can then bind to receptor proteins within the postsynaptic cell, which causes a change in the electrical state of the cell. This change in electrical state can either excite the cell, passing along the chemical message, or inhibit it. Excess serotonin molecules are taken back up by the presynaptic cell and reprocessed.
into the synaptic cleft. The serotonin molecules can then bind to receptor proteins within the postsynaptic cell, which causes a change in the electrical state of the cell. This change in electrical state can either excite the cell, passing along the chemical message, or inhibit it. Excess serotonin molecules are taken back up by the presynaptic cell and reprocessed. worthlessness, insomnia and fatigue. The most concrete evidence for the connection between serotonin and depression is the decreased concentrations of serotonin metabolites in the cerebrospinal fluid and brain tissues of depressed people.
worthlessness, insomnia and fatigue. The most concrete evidence for the connection between serotonin and depression is the decreased concentrations of serotonin metabolites in the cerebrospinal fluid and brain tissues of depressed people. serotonin from sending neural messages in the brain. Once the LSD molecule is bound to the receptor proteins the message is not carried any further. Instead the impulse is redirected to the older parts of the brain, where the bloodstream then takes it to the sense interpretive centres and the motor areas.
serotonin from sending neural messages in the brain. Once the LSD molecule is bound to the receptor proteins the message is not carried any further. Instead the impulse is redirected to the older parts of the brain, where the bloodstream then takes it to the sense interpretive centres and the motor areas.



















































 Acetate
Acetate









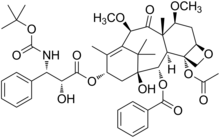
 Ridaforolimus
Ridaforolimus
































 300
300 


