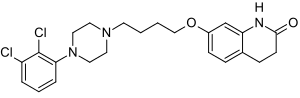
Aripiprazole
7-[4-[4-(2,3-dichlorophenyl)-1- piperazinyl]butoxy]- 3,4-dihydro-2(1H)-quinolinone.
END AUG 2014
The US Food and Drug Administration (FDA) has received a new drug application (NDA) from Ireland-based Alkermes for its aripiprazole lauroxil to treat schizophrenia.
Aripiprazole lauroxil is an injectable atypical antipsychotic with one-month and two-month formulations, developed for the treatment of schizophrenia, which is a chronic, severe and disabling brain disorder.
The company has submitted the application based on positive results from the pivotal phase three study that assessed the efficacy and safety of aripiprazole lauroxil, where the drug demonstrated significant improvements in schizophrenia symptoms when compared to a placebo.
Alkermes CEO Richard Pops said: “We have designed aripiprazole lauroxil to be a differentiated treatment option for schizophrenia, with a ready-to-use format with multiple dosing options, to help meet the individual needs of patients and their healthcare providers.
“These attributes, together with the robust clinical data observed in the pivotal study, position aripiprazole lauroxil to be a meaningful new entrant in the growing long-acting injectable antipsychotic market, and we look forward to working with the FDA to bring this important new medication to patients and physicians as quickly as possible.”
The study, in which both doses of aripiprazole lauroxil tested, including 441mg and 882mg, reached the primary endpoint with statistically significant and clinically meaningful reductions in positive and negative syndrome scale (PANSS) scores, according to the company.
In addition, it met all secondary endpoints and demonstrated significant improvements in schizophrenia symptoms against the placebo.
- ALKS 9070
- ALKS 9072
- Aripiprazole lauroxil
- RDC 3317
- RDC-3317
- UNII-B786J7A343
Aripiprazole lauroxil [USAN] CAS 1259305-29-7

| Systematic (IUPAC) name | |
|---|---|
| 7-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butoxy}-3,4-dihydroquinolin-2(1H)-one | |
| Clinical data | |
| Trade names | Abilify |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603012 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | B3 (AU) C (US) |
| Legal status | Prescription Only (S4) (AU) ℞-only (CA) POM (UK) ℞-only (US) |
| Routes | Oral (via tablets, orodispersable tablets, and oral solution); intramuscular (including as a depot) |
| Pharmacokinetic data | |
| Bioavailability | 87%[1][2][3][4] |
| Protein binding | >99%[1][2][3][4] |
| Metabolism | Hepatic (liver; mostly via CYP3A4 and CYP2D6[1][2][3][4]) |
| Half-life | 75 hours (active metabolite is 94 hours)[1][2][3][4] |
| Excretion | Renal (27%; <1% unchanged), Faecal (60%; 18% unchanged)[1][2][3][4] |
| Identifiers | |
| CAS number | 129722-12-9 |
| ATC code | N05AX12 |
| PubChem | CID 60795 |
| IUPHAR ligand | 34 |
| DrugBank | DB01238 |
| ChemSpider | 54790 |
| UNII | 82VFR53I78 |
| KEGG | D01164 |
| ChEBI | CHEBI:31236 |
| ChEMBL | CHEMBL1112 |
| Chemical data | |
| Formula | C23H27Cl2N3O2 |
| Mol. mass | 448.385 |
Aripiprazole (/ˌɛərɨˈpɪprəzoʊl/ AIR-i-PIP-rə-zohl; brand names: Abilify, Aripiprex) is a partial dopamine agonist of the second generation (or atypical) class of antipsychotics that is primarily used in the treatment of schizophrenia, bipolar disorder, major depressive disorder (as an add on to other treatment), tic disorders, and irritability associated with autism.[5]
It was approved by the U.S. Food and Drug Administration (FDA) for schizophrenia on November 15, 2002 and the European Medicines Agency on 4 June 2004; for acute manic and mixed episodes associated with bipolar disorder on October 1, 2004; as an adjunct for major depressive disorder on November 20, 2007;[6] and to treat irritability in children with autism on 20 November 2009.[7] Likewise it was approved for use as a treatment for schizophrenia by the TGA of Australia in May 2003.[1]
Aripiprazole was developed by Otsuka in Japan, and in the United States, Otsuka America markets it jointly with Bristol-Myers Squibb.
Regulator status
In the United States, the FDA has approved aripiprazole for the treatment of schizophrenia in adults and adolescents (aged 13–17), of manic and mixed episodes associated with Bipolar I (One) Disorder with or without psychotic features in adults, children and adolescents (aged 10–17),[59] of irritability associated with autism in pediatric patients (aged 6–17),[60] and of depression when used along with antidepressants in adults.[61]
Aripiprazole has been approved by the FDA for the treatment of acute manic and mixed episodes, in both pediatric patients aged 10–17 and in adults.[62]
In 2007, aripiprazole was approved by the FDA for the treatment of unipolar depression when used adjunctively with an antidepressant medication.[63] It has not been FDA-approved for use as monotherapy in unipolar depression.
Patent status
Otsuka’s US patent on aripiprazole expires on October 20, 2014;[64] however, due to a pediatric extension, a generic will not become available until at least April 20, 2015.[62] Barr Laboratories (now Teva Pharmaceuticals) initiated a patent challenge under the Hatch-Waxman Act in March 2007.[65] On November 15, 2010, this challenge was rejected by a United States district court in New Jersey.[1][2]
Dosage forms

Abilify 2mg tablets (US)
- Intramuscular injection, solution: 9.75 mg/mL (1.3 mL)
- Solution, oral: 1 mg/mL (150 mL) [contains propylene glycol, sucrose 400 mg/mL, and fructose 200 mg/mL; orange cream flavor]
- Tablet: 2 mg, 5 mg, 10 mg, 15 mg, 20 mg, 30 mg
- Tablet, orally disintegrating: 10 mg [contains phenylalanine 1.12 mg; creme de vanilla flavor]; 15 mg [contains phenylalanine 1.68 mg; creme de vanilla flavor]
Synthesis
Aripiprazole can be synthesized beginning with a dichloroaniline and bis(2-chloroethyl)amine:[66]
- U.S. Patent No.4, 734, 416 and U.S. Patent No.5,006,528 discloses the Aripiprazole, 7-{4- [4- (2, 3-dichlorophenyl) -1-piperazinyl] butoxy}- 3,4-dihydro-2 (IH) -quinolinone or 7-{4-[4- (2, 3-dichlorophenyl) -1- piperazinyl] butoxy}-3, 4-dihydro carbostyril, is a typical antipsychotic agent useful for the treatment of Schizophrenia, having the formula as given below.

Aripiprazole
U.S. patent No.5,006,528 discloses preparation of Aripiprazole and its pharmaceutically acceptable acid-addition salts. The process for the preparation of acid salts involves reaction of Aripiprazole with a pharmaceutically acceptable inorganic acids such as hydrochloric acid, sulfuric acid, phosphoric acid, hydrobromic acid, and the like; organic acids such as oxalic acid, maleic acid, fumaric acid, maleic acid, tartaric acid, citric acid, . benzoic acid and the like as per Scheme-1. Scheme- 1

a. K2CO3, Water K CH2CI2 c. Column chromatographic purification d. n-Hexane – Ethaπol

ARIPIPRAZOLE ACID SALT
The product Aripiprazole .obtained by the above process has melting point of 139.0° – 139.5°C.
The process involves purification of the intermediate, 7-(4- bromobutoxy) -3, 4-dihydrocarbostyril (III) by silica gel column chromatography to remove impurities formed during the reaction. The process further involves two recrystallizations of Aripiprazole from ethanol to obtain the pure Aripiprazole though compromising on yields by increasing the operational cost of the product. PCT publication WO 03/026659 discloses low hygroscopic forms of
Aripiprazole and the process for their preparation from the Aripiprazole hydrate Form SA’ . It further states that the anhydrous
Aripiprazole made by the Japanese patent publication No. 191256/1990, yields the Aripiprazole, which is significantly hygroscopic. As per PCT publication WO 03/026659 anhydrous crystals of Aripiprazole exist as type-I crystals and type-II crystals. Further discloses that the type-I crystals are prepared -by recrsytallization from ethanol solution of
Aripiprazole or by heating Aripiprazole hydrate at 800C and type-II crystals by heating type-I crystals at 130 to 1400C for 15 hrs.
PCT application Publication WO 03/026659 discloses process for the Aripiprazole polymorphic form-B by heating the Aripiprazole hydrate
‘A’ at 90 – 125°C for about 3 – 50 hrs. The process for Polymorphic
Form-C is by heating the Aripiprazole anhydrous to a temperature of 140
– 1500C. The process for Form-D is recrystallization from toluene; process for Form-E is heating with acetonitrile or by recrystallization from acetonitrile and the process for Form-F is by heating the suspension of anhydrous Aripiprazole in acetone. The polymorphic Form-G is by heating to 1700C for at least 2 weeks in a sealed tube, which is a glassy mass.
PCT publication WO 03/026659 further discloses the characterization data X-ray diffraction pattern; IR absorption and DSC of Form B, Form C, Form-D, Form-E, Form-F and Form-G.It further reported the melting point of Aripiprazole anhydrous Form B as 139.7°C-






Research
Perhaps owing to its mechanism of action relating to dopamine receptors, there is some evidence to suggest that aripiprazole blocks cocaine-seeking behavior in animal models without significantly affecting other rewarding behaviors (such as food self-administration).[67] Aripiprazole may be counter-therapeutic as treatment for methamphetamine dependency because it increased methamphetamine’s stimulant and euphoric effects, and increased the baseline level of desire for methamphetamine.[68]
http://www.google.com/patents/WO2006030446A1?cl=en

Scheme-3
Aripiprazole Acid addition salt

Form-A, B, C , D , E , F Type-I & Type-II Aripiprazole acid salts used for the preparation of polymorphs
…………………………….
| United States | 5006528 | 1994-10-20 | 2014-10-20 |
| United States | 7115587 | 2005-01-21 | 2025-01-21 |
Aripiprazole, 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro carbostyril or 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-butoxy}-3,4-dihydro-2 (1H)-quinolinone, is an atypical antipsychotic agent useful for the treatment of schizophrenia (U.S. Pat. No. 4,74,416 and U.S. Pat. No. 5,006,528). Schizophrenia is a common type of psychosis characterized by delusions, hallucinations and extensive withdrawal from others. Onset of schizophrenia typically occurs between the age of 16 and 25 and affects 1 in 100 individuals worldwide. It is more prevalent, than Alzheimer’s disease, multiple sclerosis, insulin-dependent diabetes and muscular dystrophy. Early diagnosis and treatment can lead to significantly improved recovery and outcome. Moreover, early therapeutic intervention can avert costly hospitalization.
Aripiprazole (Aripiprazole) is an atypical antipsychotic, on 15 November 2002 by the U.S. FDA clearance to market, its efficacy is through the dopamine D2 receptor and serotonin 5HT1A receptor partial agonist activity and serotonin 5HT2A receptor antagonism activity mediated common. With its unique mechanism of action and safety assessment, aripiprazole known as third-generation antipsychotic drugs.
[0003] Aripiprazole is a quinolinone derivative, developed by the Japanese company Otsuka Pharmaceutical, the chemical name
Is: 7 – {4 – [4 – (2,3 – dichlorophenyl)-1_ piperazinyl] butoxy} -3,4 – dihydro-quinolone, the following structural formula:
[0004]

[0005] For the preparation of aripiprazole, Japanese OtsukaPharmaceutical’s patent EP 0367141A2, and related patents US4234585, CN89108934 preparation methods described in 5. In addition, the patent CN1450056A, CN1562973A, CN1784385A, CN1680328A, CN1576273A, etc. describe some of these five Preparation
Method is very similar way. These preparation methods are direct or indirect use of 7 – hydroxy -3,4 – dihydro – quinolin-2 – one (HCS) that the key to higher prices of raw materials, and some methods involve harsh reaction conditions, poor selectivity, low yield, but also increases the cost of industrial production of the product.
[0006] Chinese patent CN1304373C preparation method is not described in the 7 – hydroxy-3 ,4 _ dihydro-2_ (1H) – quinoline
Quinolone intermediates for their preparation of the core reaction is as follows:
[0007]

[0008] This reaction is Friedel-Crafts alkylation reaction, there is a harsh reaction conditions, the yield is low, the reaction selectivity is poor, the shortcomings of high emissions, is not conducive to industrial mass production. SUMMARY OF THE INVENTION
[0009] In order to solve the above problems, the present invention provides a simple, high selectivity, high yield, low cost, environmentally friendly, easy to prepare industrialization aripiprazole and intermediates thereof.
[0010] The technical solution of the present invention, the present invention provides in one aspect a process for preparingaripiprazole novel intermediates.
[0011] The present invention, on the other hand provides a method for the preparation of intermediates.
[0012] The present invention provides the use of the other intermediates for preparing aripiprazole two new preparation methods.
[0013] Specifically, the present invention relates to novel intermediates, compounds of formula ⑴:
[0014]

[0015] wherein, R is selected from methyl, ethyl, propyl, isopropyl, butyl, t-butyl, benzyl and other common alkyl groups in any one, and preferably is ethyl.
[0016] Compound of formula ⑴: 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate, is the following prepared by the procedure:
[0017] Step one, the acylation reaction: with 4 – methyl – 3 – nitro-phenol (VIII) and acetic anhydride as the raw material, DMAP as catalyst, to give 4 – methyl – 3 – nitrophenyl acetate ( VII).
[0018] wherein 4 – methyl – 3 – nitro-phenol (VIII), acetic anhydride, DMAP molar ratio is preferably 1: 1.0 to 1.4: 0.05, at room temperature, the reaction time is preferably 0.5 to 3 hours.
[0019] Step two, the bromination reaction: The resulting product, 4 to Step one – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide and benzoyl peroxide as a raw material , carbon tetrachloride solvent reflux, to give 4 – bromomethyl-3 – nitrophenyl acetate (VI).
[0020] wherein 4 – methyl – 3 – nitrophenyl acetate (VII), N-bromosuccinimide, benzoyl peroxide molar ratio is preferably 1: 1 to 1.2: 0.05, reaction time is preferably 4-18 hours.
[0021] Step three, instead of the reaction: in an appropriate solvent, adding an alkaline agent and diethyl malonate was stirred in an ice bath, was added dropwise step two the resulting product, 4 – bromomethyl-3 – nitrophenyl yl acetate (VI) solution after completion of the addition reaction of 1 to 3 hours to obtain a brown liquid product, 2 – (4_ acetoxy-2 – nitrobenzyl) malonate (V).
[0022], wherein the alkali agent is a common organic or inorganic base selected from sodium methoxide, sodium ethoxide, sodium hydride, sodium tert-butoxide or potassium tert-butoxide, preferably sodium tert-butoxide; the solvent is selected from tetrahydrofuran, methanol, ethanol, butanol, tert-butanol, toluene or N, N-dimethylformamide; 4 – bromomethyl-3 – nitrophenyl acetate (VI), alkaline agent and lipid diethyl molar ratio is preferably 1: 1.0 to 1.8: 1.0 to 1.4.
[0023] Step 4 Hydrolysis decarboxylation: the product obtained in Step Three 2 – (4_ acetoxy-2 – nitro-benzyl)-malonic acid diethyl ester (V) was added concentrated hydrochloric acid and a suitable solvent, heating and stirring reflux, to give a yellow solid product 3 – (4_ hydroxy-2 – nitrophenyl) propionic acid (IV).
[0024] wherein the solvent is selected from water, methanol, ethanol or acetic acid, water soluble solvent, was heated with stirring under reflux time is preferably 3 to 18 hours. [0025] Step five, the esterification reaction: the product obtained in step 4, 3 – (4 – hydroxy-2 – nitrophenyl) propionic acid (IV) was added to an appropriate solvent, the mixture was stirred in an ice bath, was added dropwise thionyl sulfone, after completion of the addition reaction of 1 to 3 hours, to give a pale brown liquid product 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III).
[0026] wherein the solvent is selected from anhydrous methanol, ethanol, propanol, isopropanol, butanol, t-butanol, benzyl alcohol, alcohol and other common solvents.
[0027] Step VI substitution reaction: 1,4 – dibromobutane was added to an appropriate solvent and an alkaline reagent, heated to 50 ~ 100 ° C, the product obtained was added dropwise Step Five 3 – (4_ hydroxy – nitrophenyl) propionate (III) solution, after the addition was complete the reaction was kept 2 to 4 hours to obtain a brown liquid product 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II).
[0028] wherein the solvent is selected from methanol, 95% ethanol, ethanol, acetonitrile and N, N-dimethylformamide, and the like; said alkaline agent is a common organic or inorganic weak base, such as triethylamine, pyridine, potassium carbonate, sodium carbonate, etc..
[0029] Step 7 condensation reaction: the product obtained in Step Six 3 – (4 – (4 – bromo-butoxy)-2 – nitrophenyl) propionate (II) adding a suitable solvent, (2,3 – dichlorophenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, to obtain
The intermediate product 3 – (4 – (4 – (4 – (2,3 – dichlorophenyl)-piperazin-1 – yl) butoxy)-2 – nitrophenyl) propionate ⑴.
[0030] Among them, 3 – (4 – (4 – (4 – (2,3 _-dichlorophenyl)-piperazinyl) butoxy) _2_ nitrophenyl) propionate (I), (2, 3 – dichloro-phenyl)-piperazine hydrochloride 1_, alkaline reagents and catalysts, the four molar ratio is preferably 1: 0.9 to 1.0: 2.0 to 2.2: 0.05 to 0.5. The solvent is selected from methanol, ethanol and N, N-dimethylformamide, acetonitrile and the like. Step six of the alkaline reagent and alkaline reagent used in the same, said catalyst is a common low-iodine salts, such as sodium iodide, potassium iodide.
[0031] The present invention provides two other hand, the use of a compound of formula ⑴ preparing aripiprazole new method.
[0032] Method one: ⑴ intermediate compound of formula in an appropriate solvent in the acid or salt or a base in the presence of a reducing agent under the action of restoring ring closure reaction to obtain aripiprazole.
[0033] Method one reductive cyclization of the reducing agent used is iron, zinc, sodium sulfide, stannous chloride, and preferably iron; reaction solvent is selected from water, methanol, ethanol, ethyl acetate or in one or more of the mixed solvent; said acid is a common organic or inorganic acid, preferably acetic acid or hydrochloric acid; said salt is a common inorganic or organic salts selected from chloride, ferrous chloride, , ammonium sulfate, calcium chloride, zinc chloride, sodium chloride, sodium bromide or sodium acetate and the like; common said base is an inorganic base selected from sodium hydroxide, potassium hydroxide or sodium bicarbonate; the reduction ring-closing reaction temperature range of 30 ~ 140 ° C, preferably about 80 ° C; reaction time ranges from about 0.5 to 8 hours, preferably 2 hours.
[0034] Method two: ⑴ intermediate compound of formula in an appropriate solvent in the first catalyst, the reduction reaction, and then carried out in a suitable solvent can be prepared by cyclization of aripiprazole.
[0035] The reduction reaction of the second approach, the reducing agent is hydrogen or a carboxylic acid; the catalyst is selected from molybdenum, molybdenum dioxide or Raney nickel, preferably Raney nickel; the solvent is selected from methanol, ethanol, ethyl acetate or acetic acid, preferably ethanol; said ring-closing reaction of the solvent is selected from N, N-dimethylformamide, trichlorobenzene or xylene; reaction temperature range of 50 ~ 180 ° C, preferably about 70 ~ 150 ° C; reaction time the range of about 1 to 8 hours.
[0036] In summary, the present invention is described for preparing aripiprazole method in 4– methyl – 3 – nitro-phenol (VIII) as a starting material, by acetylation protected hydroxy, radical instead of 4 – bromomethyl-3 – nitrophenyl acetate (VI), the diethyl malonate and a nucleophilic substitution reaction to obtain 2 – (4_ acetoxy-2 – nitrobenzyl ) malonic acid diethyl ester (V), which is decarboxylated by hydrolysis, esterification, to give 3 – (4 – hydroxy-2 – nitrophenyl) propionate (III), the reaction product with dibromobutane an ether compounds, and with (2,3 – dichlorophenyl)-piperazine hydrochloride 1_ condensation, to give 3 – (4 – (4 – (4 – (2,3 – dichlorophenyl) piperazine -1 – yl) butoxy) -2 – nitrophenyl) propionate (I), and then by reductive cyclization step, or first reduced and then ring-closing reaction of aripiprazole. The synthetic route of the present invention is as follows: [0037]

According to Example 1 of Japanese Unexamined Patent Publication No. 191256/1990, anhydrous aripiprazole crystals are manufactured for example by reacting 7-(4-bromobutoxy)-3,4-dihydrocarbostyril with 1-(2,3-dichlorophenylpiperadine and recrystallizing the resulting raw anhydrousaripiprazole with ethanol. Also, according to the Proceedings of the 4th Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996), anhydrousaripiprazole crystals are manufactured by heating aripiprazole hydrate at 80° C. However, the anhydrous aripiprazole crystals obtained by the aforementioned methods have the disadvantage of being significantly hygroscopic.
The hygroscopicity of these crystals makes them difficult to handle since costly and burdensome measures must be taken in order ensure they are not exposed to moisture during process and formulation. Exposed to moisture, the anhydrous form can take on water and convert to a hydrous form. This presents several disadvantages. First, the hydrous forms of aripiprazole have the disadvantage of being less bioavailable and less dissoluble than the anhydrous forms ofaripiprazole. Second, the variation in the amount of hydrous versus anhydrousaripiprazole drug substance from batch to batch could fail to meet specifications set by drug regulatory agencies. Third, the milling may cause the drug substance, Conventional Anhydrous Aripiprazole, to adhere so manufacturing equipment which may further result in processing delay, increased operator involvement, increased cost, increased maintenance, and lower production yield. Fourth, in addition to problems caused by introduction of moisture during the processing of these hygroscopic crystals, the potential for absorbance of moisture during storage and handling would adversely affect the dissolubility of aripiprazole drug substance. Thus shelf-life of the product could be significantly decreased and/or packaging costs could be significantly increased. It would be highly desirable to discover a form of aripiprazole that possessed low hygroscopicity thereby facilitating pharmaceutical processing and formulation operations required for producing dosage units of an aripiprazole medicinal product having improved shelf-life, suitable dissolubility and suitable bioavailability.
Also, Proceedings of the 4 the Japanese-Korean Symposium on Separation Technology (Oct. 6-8, 1996) state that, anhydrous aripiprazole crystals exist as type-I crystals and type-II crystals; the type-I crystals of anhydrous aripiprazolecan be prepared by recrystallizing from an ethanol solution of aripiprazole, or by heating aripiprazole hydrate at 80° C.; and the type-II crystals of anhydrousaripiprazole can be prepared by heating the type-I crystals of anhydrousaripiprazole at 130 to 140° C. for 15 hours.
By the aforementioned methods, anhydrous aripiprazole type-II crystals having high purity can not be easily prepared in an industrial scale with good repeatability.
Chemical Synthesis of Aripiprazole (active ingredient for Abilify)

Experimental Procedures for the preparation of Aripiprazole (Abilify, aripiprazole)
US 5,006,528 discloses process for the preparation of Aripiprazole in two steps The first step comprises synthesis of 7 -. (4-bromobutoxy) -3,4-dihydrocarbostyril (7-BBQ) by alkylating the hydroxy group of 7-hydroxy-3, 4 -dihydrocarbostyril (7-HQ) with 1 ,4-dibromobutane using potassium carbonate in water at reflux temperature for 3 hours to obtain 7-BBQ in 68% yield The resulting 7-BBQ is further reacted with 1 -. (2,3 – dichlorophenyl)-piperazine to obtain Aripiprazole.
Preparation of 7 – (4-Bromobutoxy) 3 ,4-dihydro-2 (1H) quinolinon ( 7 – (4-Bromobutoxy) 3 ,4-dihydrocarbostyril; 7-BBQ)
7-Hydroxy-3 ,4-dihydro-2 (1H)-quinolinone (aka 7-Hydroxy-3 ,4-dihydrocarbostyril, 60gm) and potassium carbonate (76.3 gm) were taken in acetonitrile (1200ml) at room temperature. To this tetra butyl ammonium iodide (13.7 gm) and 1 ,4-dibromobutane (238.5gm) were added and heated at 40 – 45 ° C for 24 hours Reaction mass was cooled upto room temperature and was filtered off The resulting filtrate was distilled off.. under vacuum. The resultant mass was cooled to 25-30 ° C and cyclohexane (300 ml) was added under stirring. The resulting solid was filtered off and was dried. The resulting solid was taken in water and was stirred for few minutes. The . solid was filtered and dried under vacuum at 55-60 ° C for 20 hours to obtain title compound mp 110.5-111 ° C; 1H NMR (DMSO-d6) ä 1.81 (2H, m,-CH2-), 1.95 (2H , m,-CH2-), 2.41 (2H, t, J) 7 Hz,-CH2CO-), 2.78 (2H, t, J) 7 Hz,-CH2-C-CO-), 3.60 (2H, t, J) 6 Hz,-CH2Br), 3.93 (2H, t, J) 6 Hz, O-CH2-), 6.43 (1H, d, J) 2.5 Hz), 6.49 (1H, dd, J) 2.5, 8 Hz ), 7.04 (1H, d, J) 8 Hz), 9.98 (1H, s, NHCO). Anal. (C13H16NO2Br) C, H, N.
Yield: 73-75%; Purity: 93-95%
Preparation of Aripiprazole (7 – {4 – [4 – (2,3-Dichlorophenyl) piperazin-1-yl] butoxy} 3 ,4-dihydroquinolin-2 (1H)-One)
7 – (4-Bromobutoxy)-l ,2,3,4-tetrahydroquinolin-2-one (50 gm) was taken in acetonitrile (500 ml) at 25-30 ° C. To this potassium carbonate (67.2 gm) and l – (2,3 – dichlorophenyl). piperazine hydrochloride (44.9gm) were added under stirring The reaction mixture was refluxed at 80-85 ° C for 8 hours The reaction mass was cooled to room temperature, filtered and the resulting solid was washed. with acetonitrile. To the resulting solid, water was added and was stirred. The solid was filtered off, washed with water and dried under vacuum at 75-80 ° C for 15 hrs. The resulting crude aripiprazole was crystallized from isopropyl alcohol and water to . obtain title compound Yield: 75-80%; Dimer Impurity: <0.1% 1H NMR:. DMSO-d6 d 9.96 [1H, s, NH]; 7.29 [2H, m, Ar]; 7.13 [1H, q, Ar ]; 7.04 [1H, d, Ar]; 6.49 [1H, dd, Ar]; 6.45 [1H, d, Ar]; 3.92 [2H, t,-CH2-O-]; 2.97 [4H, bb, 2 ( -CH2-)]; 2.78 [2H, t,-CH2-N2-)]; 2.39 [4H, m, 2 (-CH2-)]; 1.73 [2H, m, – CH2-]; 1.58 [2H, m .,-CH2-] IR: cm-1 3193; 2939; 2804; 1680; 1627; 1579; 1520; 1449; 1375; 1270; 1245; 1192; 1169; 1045; 965; 649; 869; 780; 712; 588 .
For the Process of references Aripiprazole (Abilify, Japanese: Oh, Bldg re phi, Ann reピplastic AKZO have suitable; Chinese: Ann-law who, aripiprazole)
Yasuo Oshiro, Seiji Sato, Nobuyuki Kurahashi, Tatsuyoshi Tanaka, Tetsuro Kikuchi, Katsura Tottori, Yasufumi Uwahodo, and Takao Nishi; Novel Antipsychotic Agents with Dopamine autoreceptor Agonist Properties: Synthesis and Pharmacology of 7 – [4 – (4-Phenyl-1- piperazinyl) butoxy] – 3,4-dihydro-2 (1H)-quinolinone Derivatives ; J. Med Chem. 1998, 41, 658-667.
Yasuo Oshiro, Seiji Sato, Nobuyuki Kurahashi; Carbostyril Derivatives , Otsuka Pharmaceutical Co., Ltd.;. U.S. Patent 5006528 ; Issue Date: Apr 9, 1991
BANDO, Takuji, YANO, Katsuhiko, FUKANA, Makoto, AOKI, Satoshi; Method for producing fine particles of aripiprazole anhydride crystals b; OTSUKA PHARMACEUTICAL CO, LTD, WO 2013002420 A1..
Yuanqiu Hui, Chen Hongwen, Qian Wen, firewood rain column, Xu Dan, Yang Zhimin, Tian Zhoushan; method for preparing high purity of aripiprazole; NJCTT Pharmaceutical Co., Ltd.; application number: 201210292382.0; Publication Number: CN102863377A; Publication date: 2013.01.09 After (The invention relates to the field of medicine and chemical industry, in particular to a method for preparing high purity of aripiprazole would join aripiprazole A solvent is heated, filtered, and the filtrate was added to a solvent B, low temperature mixing, filtration, the filter cake is suspended in water, adjusted to alkaline pH of the aqueous solution, filtration, high temperature vacuum dried to obtain a high-purity refined product Aripiprazole This method is simple, high purity, suitable for the industrial the large-scale application)
ZHENG Siji, LIU Xiaoyi, FU Linyong, TAN Bo, ZHOU Min:.. ARIPIPRAZOLE MEDICAMENT FORMULATION AND PREPARATION METHOD THEREFOR / FORMULATION DE MÉDICAMENT ARIPIPRAZOLE ET SON PROCÉDÉ DE PRÉPARATION / a aripiprazole pharmaceutical formulation and preparation method SHANGHAI ZHONGXI. PHARMACEUTICAL January 2013: WO 2013/000391
Zheng Si Ji, Liu Xiaoyi, Fulin Yong, Tan Bo, Zhou Min: A aripiprazole pharmaceutical formulation and preparation method; Shanghai Pharmaceutical Co., Ltd. and Western; Publication date: 2013.01.02: Application Number: CN 201210235157.3; Publication Number: CN102846543A (the invention provides a method for preparing aripiprazole pharmaceutical formulation, comprising the steps of: an acidic solution containing aripiprazole is dissolved in the acidulant, to obtain an acidic solution containing the drug; Thereafter, the resulting drug-containing acidic solution alkalizing agents and materials prepared by wet granulation or suspension to give aripiprazole pharmaceutical formulation; said excipients include antioxidants)
Zheng Si Ji; Tan wave; Fulin Yong; Liu Xiaoyi; Yuanshao Qing; Cao Zhihui; aripiprazole Ⅰ type microcrystalline, aripiprazole solid preparation and preparation methods; application number: 201110180032.0; Publication Number: CN102850268A; Publication Date: 2013.01.02
Cai Fu Bo, Qin Xinrong, Du Xiaochun, Li Ling; kind of aripiprazole improved method of synthesis; Chengdu Nakasone Pharmaceutical Group Co., Ltd.; Application Number: 200910058148.X; Publication Number: CN101781246A; Publication date: 2010.07.21 (the invention provides a method of synthesis of aripiprazole improved method according to the modified method of the present invention, aripiprazole into the etherification reaction and condensation reaction of two-step synthesis, by an etherification reaction in the quinolone compound and at least 6-fold molar equivalents of 1,4 – dihalo-butane reacted with a non-polar solvent ether aripiprazole precipitate, and recovering 1,4 – dihalo-butane recycling; azeotropic condensation reaction of a ketone to be / water mixture as solvent, aripiprazole etherified with a piperazine compound or a salt thereof in the presence of a base under reflux and alkaline metal iodide compound conditions, the amount of water added to the end of the reaction, cooling crystallization, filtration, and dried to give aripiprazole. improved high yield synthesis of high purity, step simple, low cost, suitable for industrial production.)
GUPTA, Vijay Shankar, KUMAR, Pramod, VIR, Dharam; Process for producing aripiprazole in anhydrous type i crystals; JUBILANT LIFE SCIENCES LIMITED; WO 2012131451 A1
SRIVASTAVA JAYANT GUPTA Vijay Shankar;. Improved process for the preparation of 7 (4-bromobutoxy) 3,4-dihydrocarbostyril, a precursor of aripiprazole; wo2011030213 A1
No Generic Abilify in the US until April 2015
On May 7, 2012, The US Court of Appeals for the Federal Circuit ruled in favor of Otsuka Pharmaceutical Co., Ltd. In its patent litigation against several companies including Israel-based Teva and Weston, Ontario-based Apotex seeking FDA approval to market generic copies of Abilify ®.. The Federal Circuit Affirmed a Decision of the U.S. District Court for the District of New Jersey Holding that the asserted claims ofU.S. Patent No. 5,006,528 Covering aripiprazole, the active Ingredient in Abilify ®, are Valid, THUS Maintaining Patent and Regulatory Protection for Abilify ® in the U.S. until at least April 20, 2015 . The Case is Otsuka Pharma Co.. V. sand Inc.., 2011-1126 and 2011-1127, US Court of Appeals for the Federal Circuit (Washington). The lower court case is Otsuka Pharmaceutical Co. v. Sandoz Inc., 07cv1000, US District Court for the District of New Jersey (Trenton).
Chemical Name for Aripiprazole (Abilify for active Ingredient): 7 – {4 – [4 – (2,3-Dichlorophenyl) piperazin-1-yl] butoxy} 3 ,4-dihydroquinolin-2 (1H)-One
CAS Number 129722 -12-9
aripiprazole chemical name 7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro-2 ( 1H) – quinolinone
Aripiprazole (, Aripiprazole, Abilify) is an atypical antipsychotic medication for the quinoline derivatives, aripiprazole is a dopamine system stabilizer first, positive and schizophrenia negative symptoms have a significant effect. For the treatment of schizophrenia, the development of Otsuka Pharmaceutical Co., Ltd., in November 15, 2002 by the U.S. Food and Drug Administration (FDA) approval in the U.S., domestic aripiprazole has (Booz clear (brisking, manufacturers : Chengdu Nakasone Pharmaceutical), Austrian (Manufacturer: Shanghai Pharmaceutical Co., Ltd. and Western)) have been approved by the listing in China. On sale in the United States where the law by Bristol-Myers Squibb is responsible. An law where the main patent protection in the United States, and more than three-quarters of its sales from the U.S., patent will expire in April 2015.
Aripiprazole synthetic route
7 – hydroxy-3 ,4. Dihydro -2 (1H) – quinolinone as a starting material, 1,4. Dibromobutane ether to give 7 – (4 – Bromo-butoxy) -3,4 – dihydro – 2 (1H) quinolinone, and then with 1 – (2,3 – dichlorophenyl) piperazine acid condensation aripiprazole (7 – [4 – [4 – (2,3 – dichlorophenyl) -1 – piperazinyl] butoxy] -3,4 – dihydro -2 (1H) – quinolinone)
Aripiprazole preparation method
7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone
A reaction flask was added 7 – hydroxy – 3,4 – dihydro -2 (1H) – quinolone 32.6 g (0.2mol), 1,4 – dibromo butane 129.5g (0.6mol), 11.2% KOH solution 250ml (0.5mol) and DMF975ml, was heated to 60 º C for 2h diluted with 1L water, the aqueous layer with ethyl acetate. acetate (300ml × 2) and the combined organic layers were washed with water, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to recover the solvent, the residue was recrystallized from isopropanol, to give 7 – (4 – Bromo-butoxy) – 3,4 – dihydro -2 (1H) – quinolone 38.7g, yield 68%, mp108 ~ 110 º C.
Synthesis of aripiprazole
in the reaction flask was added 7 – (4 – Bromo-butoxy) -3,4 – dihydro -2 (1H) – quinolone, 29.8g (0.1mol), KI25g (0.15mol) 95% Ethanol 596ml, stirred and heated to 60 º C, was added N-2 30min after 3 – dichlorophenyl piperazine 23.1g (0.1mol) and triethylamine 20ml (0.15mol), stirred for 8h at 60 º C the mixture is filtered. crystallization filtrate was cooled, filtered and the filter cake was recrystallized twice from ethanol and dried to obtain aripiprazole 25.6g, yield 57%, mp138.9 ~ 139.6 º C.
References
- “Product Information for ABILIFYTM Aripiprazole Tablets & Orally Disintegrating Tablets”. TGA eBusiness Services. Bristol-Myers Squibb Australia Pty Ltd. 1 November 2012. Retrieved 22 October 2013.
- “ABILIFY (aripiprazole) tablet ABILIFY (aripiprazole) solution ABILIFY DISCMELT (aripiprazole) tablet, orally disintegrating ABILIFY (aripiprazole) injection, solution [Otsuka America Pharmaceutical, Inc.]”. DailyMed. Otsuka America Pharmaceutical, Inc. April 2013. Retrieved 22 October 2013.
- “Abilify Tablets, Orodispersible Tablets, Oral Solution – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Otsuka Pharmaceuticals (UK) Ltd. 20 September 2013. Retrieved 22 October 2013.
- “ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS”. European Medicines Agency. Otsuka Pharmaceutical Europe Ltd. Retrieved 22 October 2013.
- http://www.webmd.com/drugs/drug-64439-Abilify+Oral.aspx?drugid=64439&drugname=Abilify+Oral&source=1
- Hitti, Miranda (20 November 2007). “FDA OKs Abilify for Depression”. WebMD. Archived from the original on 5 December 2008. Retrieved 8 December 2008.
- Keating, Gina (23 November 2009). “FDA OKs Abilify for child autism irritability”. Reuters. Retrieved 22 September 2010.
- “abilify”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- Belgamwar RB, El-Sayeh HG (Aug 10, 2011). “Aripiprazole versus placebo for schizophrenia.”. The Cochrane database of systematic reviews (8): CD006622. PMID 21833956.
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, Samara M, Barbui C, Engel RR, Geddes JR, Kissling W, Stapf MP, Lässig B, Salanti G, Davis JM (2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ (2013). “World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects”. World J. Biol. Psychiatry 14 (1): 2–44. doi:10.3109/15622975.2012.739708. PMID 23216388.
- El-Sayeh HG, Morganti C (Apr 19, 2006). “Aripiprazole for schizophrenia.”. Cochrane database of systematic reviews (Online) (2): CD004578. doi:10.1002/14651858.CD004578.pub3. PMID 16625607.
- Belgamwar RB, El-Sayeh HG (Aug 10, 2011). “Aripiprazole versus placebo for schizophrenia.”. Cochrane database of systematic reviews (Online) (8): CD006622. doi:10.1002/14651858.CD006622.pub2. PMID 21833956.
- Bhattacharjee J, El-Sayeh HG (Jan 23, 2008). “Aripiprazole versus typicals for schizophrenia.”. Cochrane database of systematic reviews (Online) (1): CD006617. doi:10.1002/14651858.CD006617.pub2. PMID 18254107.
- Khanna P, Komossa K, Rummel-Kluge C, Hunger H, Schwarz S, El-Sayeh HG, Leucht S (Feb 28, 2013). “Aripiprazole versus other atypical antipsychotics for schizophrenia.”. Cochrane database of systematic reviews (Online) 2: CD006569. doi:10.1002/14651858.CD006569.pub4. PMID 23450570.
- “Psychosis and schizophrenia in adults: treatment and management | Guidance and guidelines | NICE”. National Institute for Health and Care Excellence.
- Barnes TR (2011). “Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology”. J. Psychopharmacol. (Oxford) 25 (5): 567–620. doi:10.1177/0269881110391123. PMID 21292923.
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ (2013). “World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects”. World J. Biol. Psychiatry 14 (1): 2–44. doi:10.3109/15622975.2012.739708. PMID 23216388.
- De Fruyt J, Deschepper E, Audenaert K, Constant E, Floris M, Pitchot W, Sienaert P, Souery D, Claes S (May 2012). “Second generation antipsychotics in the treatment of bipolar depression: a systematic review and meta-analysis.”. Journal of psychopharmacology (Oxford, England) 26 (5): 603–17. doi:10.1177/0269881111408461. PMID 21940761.
- Gitlin M, Frye MA (May 2012). “Maintenance therapies in bipolar disorders.”. Bipolar disorders. 14 Suppl 2: 51–65. doi:10.1111/j.1399-5618.2012.00992.x. PMID 22510036.
- de Bartolomeis A, Perugi G (October 2012). “Combination of aripiprazole with mood stabilizers for the treatment of bipolar disorder: from acute mania to long-term maintenance.”. Expert opinion on pharmacotherapy 13 (14): 2027–36. doi:10.1517/14656566.2012.719876. PMID 22946707.
- Komossa K, Depping AM, Gaudchau A, Kissling W, Leucht S (Dec 8, 2010). “Second-generation antipsychotics for major depressive disorder and dysthymia.”. Cochrane database of systematic reviews (Online) (12): CD008121. doi:10.1002/14651858.CD008121.pub2. PMID 21154393.
- Spielmans GI, Berman MI, Linardatos E, Rosenlicht NZ, Perry A, Tsai AC (Mar 12, 2013). “Adjunctive atypical antipsychotic treatment for major depressive disorder: a meta-analysis of depression, quality of life, and safety outcomes”. Cochrane database of systematic reviews (Online) 10 (3): CD008121. doi:10.1371/journal.pmed.1001403. PMID 23554581.
- Nelson JC, Papakostas GI (2009). “Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials”. Am J Psychiatry 166 (9): 980–91. doi:10.1176/appi.ajp.2009.09030312. PMID 19687129.
- Komossa K, Depping AM, Gaudchau A, Kissling W, Leucht S (2010). “Second-generation antipsychotics for major depressive disorder and dysthymia”. Cochrane Database Syst Rev (12): CD008121. doi:10.1002/14651858.CD008121.pub2. PMID 21154393.
- Ching H, Pringsheim T (May 16, 2012). “Aripiprazole for autism spectrum disorders (ASD).”. Cochrane database of systematic reviews (Online) 5: CD009043. doi:10.1002/14651858.CD009043.pub2. PMID 22592735.
- Joint Formulary Committee. British National Formulary (BNF) 65. Pharmaceutical Pr; 2013.
- Australian Medicines Handbook 2013 [Internet]. [cited 2013 Sep 30]. Available from: http://www.psa.org.au/shop/amh
- Truven Health Analytics, Inc. DRUGDEX® System (Internet) [cited 2013 Jun 25]. Greenwood Village, CO: Thomsen Healthcare; 2013.
- “Abilify Discmelt, Abilify Maintena (aripiprazole) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 22 October 2013.
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, Samara M, Barbui C, Engel RR, Geddes JR, Kissling W, Stapf MP, Lässig B, Salanti G, Davis JM (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis.”. Lancet 382 (9896): 951–962. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Abbasian C, Power P (March 2009). “A case of aripiprazole and tardive dyskinesia”. J Psychopharmacol (Oxford) 23 (2): 214–5. doi:10.1177/0269881108089591. PMID 18515468.
- Zaidi SH, Faruqui RA (January 2008). “Aripiprazole is associated with early onset of Tardive Dyskinesia like presentation in a patient with ABI and psychosis”. Brain Inj 22 (1): 99–102. doi:10.1080/02699050701822493. PMID 18183513.
- Maytal G, Ostacher M, Stern TA (June 2006). “Aripiprazole-related tardive dyskinesia”. CNS Spectr 11 (6): 435–9. PMID 16816781.
- http://www.medicines.org.uk/EMC/pdfviewer.aspx?isAttachment=true&documentid=16161
- “ABILIFY (aripiprazole) [package insert].”. Otsuka Pharmaceutical Co, Ltd. Retrieved 18 October 2012.
- Group, BMJ, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISBN 978-0-85369-845-6. “Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.”
- Moncrieff J (Jul 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatr Scand 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655.
- R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 105-106.
- “Abilify (Aripiprazole) – Warnings and Precautions”. DrugLib.com. 14 February 2007. Archived from the original on 4 December 2008. Retrieved 8 December 2008.
- http://www.drugs.com/cons/abilify-intramuscular.html
- http://www.drugs.com/pro/abilify.html
- Starrenburg FC, Bogers JP (April 2009). “How can antipsychotics cause diabetes mellitus? Insights based on receptor-binding profiles, humoral factors and transporter proteins”. European Psychiatry 24 (3): 164–170. doi:10.1016/j.eurpsy.2009.01.001. PMID 19285836.
- “Abilify (Aripiprazole) – Clinical Pharmacology”. DrugLib.com. 14 February 2007. Retrieved 8 December 2008.
- Brunton, Laurence (2011). Goodman & Gilman’s The Pharmacological Basis of Therapeutics 12th Edition. China: McGraw-Hill. pp. 406–410. ISBN 978-0-07-162442-8.
- “PDSP Ki Database”. National Institute of Mental Health. Retrieved 30 June 2013.
- Nguyen CT, Rosen JA, Bota RG (2012). “Aripiprazole partial agonism at 5-HT2C: a comparison of weight gain associated with aripiprazole adjunctive to antidepressants with high versus low serotonergic activities”. Prim Care Companion CNS Disord 14 (5). doi:10.4088/PCC.12m01386. PMC 3583771. PMID 23469329.
- Newman-Tancredi A, Heusler P, Martel JC, Ormière AM, Leduc N, Cussac D (2008). “Agonist and antagonist properties of antipsychotics at human dopamine D4.4 receptors: G-protein activation and K+ channel modulation in transfected cells”. Int. J. Neuropsychopharmacol. 11 (3): 293–307. doi:10.1017/S1461145707008061. PMID 17897483.
- Burstein ES, Ma J, Wong S, Gao Y, Pham E, Knapp AE, Nash NR, Olsson R, Davis RE, Hacksell U, Weiner DM, Brann MR (2005). “Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist”. J. Pharmacol. Exp. Ther. 315 (3): 1278–87. doi:10.1124/jpet.105.092155. PMID 16135699.
- Davies MA, Sheffler DJ, Roth BL (2004). “Aripiprazole: a novel atypical antipsychotic drug with a uniquely robust pharmacology”. CNS Drug Rev 10 (4): 317–36. doi:10.1111/j.1527-3458.2004.tb00030.x. PMID 15592581.
- Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA, Gonzalez AM, Sibley DR, Mailman RB (1999). “Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes”. Neuropsychopharmacology 20 (6): 612–27. doi:10.1016/S0893-133X(98)00099-2. PMID 10327430.
- Burstein ES, Ma J, Wong S, Gao Y, Pham E, Knapp AE, Nash NR, Olsson R, Davis RE, Hacksell U, Weiner DM, Brann MR (December 2005). “Intrinsic Efficacy of Antipsychotics at Human D2, D3, and D4 Dopamine Receptors: Identification of the Clozapine Metabolite N-Desmethylclozapine as a D2/D3 Partial Agonist”. J Pharmacol Exp Ther 315 (3): 1278–87. doi:10.1124/jpet.105.092155. PMID 16135699.
- Jordan S, Koprivica V, Chen R, Tottori K, Kikuchi T, Altar CA (2002). “The antipsychotic aripiprazole is a potent, partial agonist at the human 5-HT1A receptor”. Eur J Pharmacol 441 (3): 137–140. doi:10.1016/S0014-2999(02)01532-7. PMID 12063084.
- Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R (2003). “Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology”. Neuropsychopharmacology 28 (8): 1400–1411. doi:10.1038/sj.npp.1300203. PMID 12784105.
- Zhang JY, Kowal DM, Nawoschik SP, Lou Z, Dunlop J (February 2006). “Distinct functional profiles of aripiprazole and olanzapine at RNA edited human 5-HT2C receptor isoforms”. Biochem Pharmacol 71 (4): 521–9. doi:10.1016/j.bcp.2005.11.007. PMID 16336943.
- Kegeles LS, Slifstein M, Frankle WG, Xu X, Hackett E, Bae SA, Gonzales R, Kim JH, Alvarez B, Gil R, Laruelle M, Abi-Dargham A (2008). “Dose–Occupancy Study of Striatal and Extrastriatal Dopamine D2 Receptors by Aripiprazole in Schizophrenia with PET and [18F]Fallypride”. Neuropsychopharmacology 33 (13): 3111–3125. doi:10.1038/npp.2008.33. PMID 18418366.
- Yokoi F, Gründer G, Biziere K, Stephane M, Dogan AS, Dannals RF, Ravert H, Suri A, Bramer S, Wong DF (August 2002). “Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride”. Neuropsychopharmacology 27 (2): 248–59. doi:10.1016/S0893-133X(02)00304-4. PMID 12093598.
- “In This Issue”. Am J Psychiatry 165 (8): A46. August 2008. doi:10.1176/appi.ajp.2008.165.8.A46.
- “Abilify Receives Approval for Expanded Use in Children, Teens”. Psych Central. Retrieved 2012-07-16.
- “Abilify Gets FDA Approval For Autism Irritability”. Furious Seasons. Retrieved 2012-07-16.
- “FDA OKs Abilify for Depression : Antipsychotic Drug Approved for Use in Addition to Antidepressants for Treating Depression”. WebMD. Retrieved 2012-07-16.
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 8 December 2008.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021436s21,021713s16,021729s8,021866s8lbl.pdfSection 2.3 pp 7-8
- US 5006528, Oshiro, Yasuo; Seiji Sato & Nobuyuki Kurahashi, “Carbostyril derivatives”, published October 20, 1989
- “Barr Confirms Filing an Application with a Paragraph IV Certification for ABILIFY(R) Tablets” (Press release). Barr Pharmaceuticals, Inc. 2007-03-20. Retrieved 2008-12-23.
- U.S. Patent 5,006,528
- Feltenstein MW, Altar CA, See RE (2007). “Aripiprazole blocks reinstatement of cocaine seeking in an animal model of relapse”. Biol. Psychiatry 61 (5): 582–90. doi:10.1016/j.biopsych.2006.04.010. PMID 16806092.
- Roache JD (2013). “Role of the human laboratory in the development of medications for alcohol and drug dependence”. In Johnson BA. Addiction medicine: science and practice. New York: Springer. p. 145. ISBN 978-1461439899.
External links
- Abilify website
- Abilify Full Prescribing Information (for Health Care Professionals)
- U.S. National Library of Medicine: Drug Information Portal – Aripiprazole
- Abilify adverse events reported to the FDA
- Mechanism of Action Of Aripiprazole
| WO2006079548A1 * | Jan 27, 2006 | Aug 3, 2006 | Sandoz Ag | Organic compounds |
| WO2006079549A1 | Jan 27, 2006 | Aug 3, 2006 | Sandoz Ag | Salts of aripiprazole |
| WO2014060324A1 | Oct 11, 2013 | Apr 24, 2014 | Sanovel Ilac Sanayi Ve Ticaret A.S | Aripiprazole formulations |
| EP1844036A1 * | Jan 27, 2006 | Oct 17, 2007 | Sandoz AG | Salts of aripiprazole |
| EP2093217A1 * | Jan 27, 2006 | Aug 26, 2009 | Sandoz AG | Polymorph and solvates of aripiprazole |
| EP2233471A1 * | Feb 6, 2009 | Sep 29, 2010 | Adamed Sp. z o.o. | A salt of 7-{4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butoxy}-3,4.dihydro-2(1h)-quinolinone with 5-sulfosalicylic acid and its preparation process |
| EP2359816A1 | Feb 8, 2011 | Aug 24, 2011 | Sanovel Ilac Sanayi ve Ticaret A.S. | Aripiprazole formulations |
| US7504504 | Dec 16, 2004 | Mar 17, 2009 | Teva Pharmaceutical Industries Ltd. | Methods of preparing aripiprazole crystalline forms |
| US7714129 | Sep 29, 2006 | May 11, 2010 | Teva Pharmaceutical Industries Ltd. | Methods of preparing anhydrous aripiprazole form II |
| US8008490 | Jan 27, 2006 | Aug 30, 2011 | Sandoz Ag | Polymorphic forms of aripiprazole and method |
| US8188076 | Feb 26, 2010 | May 29, 2012 | Reviva Pharmaceuticals, Inc. | Compositions, synthesis, and methods of utilizing arylpiperazine derivatives |
| US8207163 | May 27, 2009 | Jun 26, 2012 | Reviva Pharmaceuticals, Inc. | Compositions, synthesis, and methods of using piperazine based antipsychotic agents |
| US8247420 | May 21, 2008 | Aug 21, 2012 | Reviva Pharmaceuticals, Inc. | Compositions, synthesis, and methods of using quinolinone based atypical antipsychotic agents |
| US8431570 | May 7, 2012 | Apr 30, 2013 | Reviva Pharmaceuticals, Inc. | Methods of utilizing arylpiperazine derivatives |
| US8461154 | May 7, 2012 | Jun 11, 2013 | Reviva Pharmaceuticals, Inc. | Methods of utilizing arylpiperazine derivatives |
| US8575185 | Feb 26, 2010 | Nov 5, 2013 | Reviva Pharmaceuticals, Inc. | Compositions, synthesis, and methods of utilizing quinazolinedione derivatives |




 Ethyl phenylacetylenecarboxylate~Phenylpropiolic acid ethyl ester
Ethyl phenylacetylenecarboxylate~Phenylpropiolic acid ethyl ester




 4-Bromo-1,2-(methylenedioxy)benzene
4-Bromo-1,2-(methylenedioxy)benzene