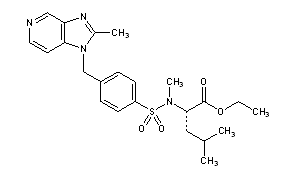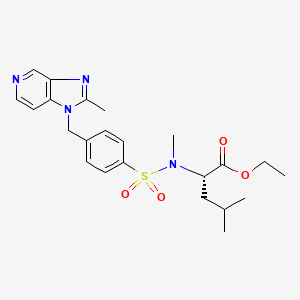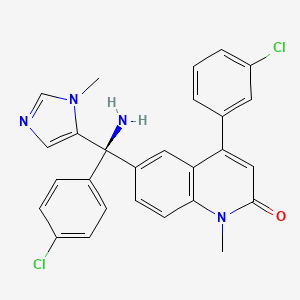
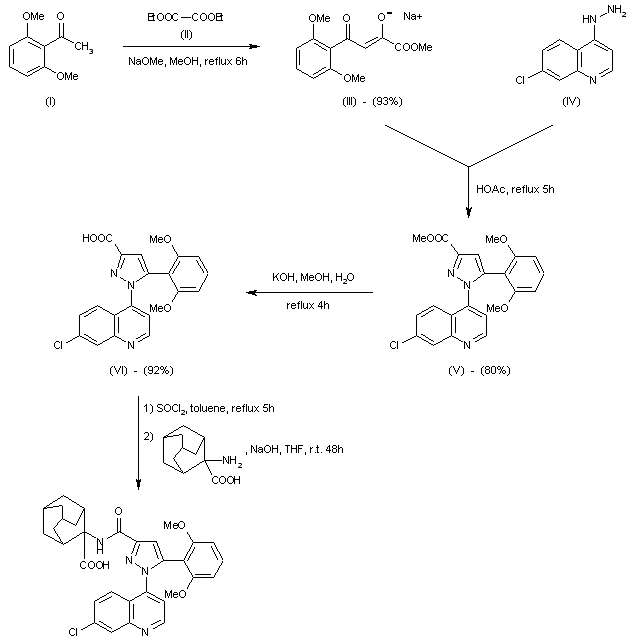
6-[(R)-Amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone;
(R)-(+)-R 115777; Zarnestra; 192185-68-5
zarnestra, 192185-72-1, R115777, R-115777, IND 58359, UNII-MAT637500A
192185-72-1, 192185-68-5
Tipifarnib (trade name Zarnestra) is a farnesyltransferase inhibitor that is being investigated in patients 65 years of age and older with newly diagnosed acute myeloid leukemia (AML). It inhibits the Ras kinase in a post translational modification step before the kinase pathway becomes hyperactive. It inhibits prenylation of the CxxX tail motif, which allows Ras to bind to the membrane where it is active. Without this step the protein cannot function.
It is also being tested in clinical trials in patients in certain stages of breast cancer.[1]
For treatment of progressive plexiform neurofibromas associated with Neurofibromatosis type I, it successfully passed phase one clinical trials but was suspended (NCT00029354) in phase two.[2][3] The compound was discovered by and is under investigation byJohnson & Johnson Pharmaceutical Research & Development, L.L.C, with registration number R115777.
Approval process
Tipifarnib was submitted to the FDA by Johnson & Johnson for the treatment of AML in patients aged 65 and over with a New Drug Application (NDA) to the Food and Drug Administration (FDA) on January 24, 2005.
In June 2005, the FDA issued a “not approvable” letter for tipifarnib.[4]
Farnesyltransferase inhibitors block the main post-translational modification of the Ras protein, thus interfering with its localization to the inner surface of the plasma membrane and subsequent activation of the downstream effectors. Although initially developed as a strategy to target Ras in cancer, farnesyltransferase inhibitors have subsequently been acknowledged as acting by additional and more complex mechanisms that may extend beyond Ras involving GTP-binding proteins, kinases, centromere-binding proteins and probably other farnesylated proteins.
A particular farnesyltransferase inhibitor is described in WO 97/21701, namely (R)-(+)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone. The absolute stereochemical configuration of the compound was not determined in the experiments described in the above-mentioned patent specification, but the compound was identified by the prefix “(B)” to indicate that it was the second compound isolated from column chromatography. The compound thus obtained has been found to have the (R)-(+)-configuration. This compound will be referred to below by its published code number R115777 and has the following formula (V).

R115777 (Tipifarnib) is a potent, orally active inhibitor of farnesylprotein transferase. It is one of the most advanced of the farnesylprotein transferase inhibitors currently reported to be in clinical development, being one of the agents that have progressed to phase III studies.
R115777 has been found to have very potent activity against neoplastic diseases. Antineoplastic activity in solid tumors, such as breast cancer, as well as in haematological malignancies, such as leukemia, have been observed. Also combination studies have been carried out demonstrating that R115777 can be safely combined with several highly active anticancer drugs.
In WO 01/53289, the racemates (±) (4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-(4-methoxy-benzylamino)-(3-methyl-3H-imidazol-4-yl)-methyl]-1-cyclopropylmethyl-1H-quinolin-2-one (racemate 1) and (±) 4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-[(4-methoxy-benzylidene)-amino]-(3-methyl-3H-imidazol-4-yl)-methyl]-1-cyclopropylmethyl-1H-quinolin-2-one (racemate 2) are prepared.

After chiral molecule separation using column chromatography, either the benzylamino or the benzilidine moiety of the resulting (+) and/or (−) enantiomers are converted to an amino group under acidic conditions.
In WO 97/21701, it is described (on page 9, line 7-14) that intermediates of formula (XIII), can be prepared by reacting an intermediate of formula (XIV), wherein W is an appropriate leaving group, such as, for example, halo, with an intermediate ketone of formula (XV). In WO 97/21701, it is described that this reaction can be performed by converting the intermediate of formula (XV) into an organometallic compound, by stirring it with a strong base such as butyl lithium and subsequently adding the intermediate ketone of formula (XV). It is further indicated that although this reaction gives at first instance a hydroxy derivative (i.e. R8 is hydroxy), said hydroxy derivative can be converted into other intermediates wherein R8 has another definition by performing art-known (functional group) transformations. The drawings of the compounds of formula (XIII), (XV) and (XIV) have been taken over from WO 97/21701 and the substituents in these drawings are as defined in WO 97/21701.

In WO 97/21701, it is also described (from page 7 line 32, to page 8 line 6) that the compounds of formula (XVI), wherein R is C1-6alkyl, R(2-8, 16-19) can be a substituent chosen from lists as defined in WO 97/21701 and R1 has a meaning as defined in WO 97/21701 apart from hydrogen, may be prepared by hydrolysing an intermediate ether of formula (XIII), according to art-known methods, such as stirring the intermediate of formula (XIII) in an aqueous acid solution. An appropriate acid can be for instance hydrochloric acid. Subsequently the resulting quinolinone, wherein R1 is hydrogen, may be transformed into a quinolinone of formula (XVI) by art-known N-alkylation. The drawings of the compounds of formula (XIII) and (XVI) have been taken over from WO 97/21701 and the substituents in these drawings are as defined in WO 97/21701.

The synthesis of R115777 as originally described in WO 97/21701, is presented in scheme 1.
Herein, in step 1, the intermediate 1-methyl imidazole in tetrahydrofuran, is mixed with a solution of n-butyllithium in a hexane solvent to which is added chlorotriethylsilane (triethylsilyl chloride), followed by a further addition of n-butyllithium in hexane, the resulting mixture being cooled to −78° C. before the addition of a solution of a compound of formula (I), i.e. 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone in tetrahydrofuran. The reaction mixture is subsequently brought to room temperature, and then hydrolysed, extracted with ethyl acetate and the organic layer worked up to obtain a compound of formula (II), i.e. (±)-6-[hydroxy(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone.
In step 2, the hydroxy compound of formula (II) is chlorinated with thionylchloride to form a compound of formula (III), i.e. (±)-6-[chloro(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone.
In step 3, the chloro compound of formula (III) is treated, with NH4OH in tetrahydrofuran to form the amino compound of formula (IV), i.e. (±)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone.
In step 4, the amino compound of formula (IV) is separated into its enantiomers by chiral column chromatography over Chiracel OD (25 cm; eluent: 100% ethanol; flow: 0.5 ml/min; wavelength: 220 nm). The pure (B)-fractions are collected and recrystallised from 2-propanol resulting in R115777, the compound of formula (V).

However, the procedure described in WO97/21701 has a number of disadvantages. For example, during the first step, the procedure results in the undesired formation of a corresponding compound of formula (XI), i.e. 6-[hydroxy(4-chlorophenyl)(1-methyl-1H-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone), in which the imidazole ring is attached to the remainder of the molecule at the 2-position of the ring, instead of the desired 5-position. At the end of the procedure, this results in the formation of a compound of formula (XII), i.e. 6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone.

Furthermore, the purification of compound (V) using chiral chromatography is expensive and disadvantageous in view of the large amounts of solvent needed and the specialised equipment required to perform a large scale chiral chromatography.
Another process for the synthesis of R115777 as described in WO 02/072574, is presented in scheme 2.
Herein, in step 1, 1-methyl imidazole in tetrahydrofuran is mixed with a solution of n-hexyllithium in a hexane solvent to which is added tri-iso-butylsilyl chloride, followed by a further addition of n-hexyllithium in hexane. The compound of formula (I) in tetrahydrofuran is then added to the reaction mixture, keeping the temperature between −5° C. and 0° C. The resulting product of formula (II) is isolated by salt formation.
In step 2, the chlorination reaction is effected by treatment of the compound of formula (II) with thionyl chloride in 1,3-dimethyl-2-imidazolidinone.
In step 3, the chloro compound of formula (III) is treated with a solution of ammonia in methanol. After the addition of water, the compound of formula (IV), precipitates and can be isolated.
In step 4, the compound of formula (IV) can be reacted with L-(−)-dibenzoyl tartaric acid (DBTA) to form the diastereomeric tartrate salt with formula (VI) i.e. R-(−)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone [R—(R*,R*)]-2,3-bis(benzoyloxy)butanedioate (2:3).
Finally, in step 5, the compound of formula (VI) is treated with aqueous ammonium hydroxide, to form the crude compound of formula (V) which is then purified by recrystallisation from ethanol to the pure compound (V).

| Patent | Submitted | Granted |
|---|---|---|
| Process for the preparation of imidazole compounds [US6844439] | 2004-07-15 | 2005-01-18 |
| TREATMENT OF MITOCHONDRIAL DISORDERS USING A FARNESYL TRANSFERASE INHIBITOR [US2010331363] | 2010-12-30 | |
| TREATMENT OF MITOCHONDRIAL DISORDERS USING A FARNESYL TRANSFERASE INHIBITOR [US2011060005] | 2011-03-10 | |
| Diastereoselective Synthesis Process with 6-Bromo-4-(3-Chlorophenyl)-2-Methoxy-Quinoline [US7572916] | 2007-12-20 | 2009-08-11 |
| Anti-cancer phosphonate analogs [US7452901] | 2006-04-13 | 2008-11-18 |
| Diastereoselective Synthesis Process for the Preparation of Imidazole Compounds [US7456287] | 2007-10-11 | 2008-11-25 |
| Diastereoselective Addition of Lithiated N-Methylimidazole on Sulfinimines [US7524961] | 2007-12-20 | 2009-04-28 |
| Therapeutic phosphonate compounds [US7645747] | 2006-11-23 | 2010-01-12 |
| TREATMENT OF PROTEINOPATHIES USING A FARNESYL TRANSFERASE INHIBITOR [US2010160372] | 2010-06-24 | |
| ANTI-CANCER PHOSPHONATE ANALOGS [US2010022467] | 2010-01-28 |

Ti(OEt)4 (0.0162 mol) was added to a mixture of (4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methanone (0.0032 mol) and (R)-(+)-2-methyl-2-propane-sulfinamide (0.0032 mol) in DCE (7 ml). The mixture was stirred and refluxed for 6 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgSO4), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/NH4OH 97/3/0.5), yielding 0.475 g of compound 25 (46%).
The compound N-[(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methylene)]-2-methyl-2-propanesulfinamide [(S(S)] can be obtained in an analogous way.
b) Preparation of N-[(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoxy-quinoline-6-yl)(1-methyl-1H-imidazole-5-yl)methyl]-2-methyl-2-propanesulfinamide [S(R)] (Compound 26)

n-Butyllithium (0.00081 mol) in hexane, was added dropwise at −78° C. to a mixture of 6-bromo-4-(3-chlorophenyl)-2-methoxy-quinoline (0.00081 mol) in THF (3 ml) under nitrogen flow. The mixture was stirred at −78° C. for 30 minutes. A solution of compound 25 (0.00065 mol) in THF (0.6 ml) was added. The mixture was stirred at −78° C. for 1 hour and 30 minutes, poured out into ice water and extracted with EtOAc. The organic layer was separated, dried (MgSO4), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm)(eluent: DCM/MeOH/NH4OH 97/3/0.1). The pure fractions were collected and the solvent was evaporated, yielding 0.138 g (36%) of compound 26, melting point 153° C.
The compound N-[(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoxy-quinoline-6-yl)(1-methyl-1H-imidazole-5-yl)methyl]-2-methyl-2-propanesulfinamide [S(S)] can be obtained in an analogous way
c) Preparation of (S)-1-(4-chlorophenyl)-1-[4-(3-chlorophenyl)-2-methoxy-quinoline-6-yl]-1-(1-methyl-1H-imidazole-5-yl)-methylamine (Compound 27)

Hydrochloric acid in isopropanol was added to a solution of compound 26 (0.000018 mol) in methanol (4.2 ml). The mixture was stirred at room temperature for 30 minutes. The mixture was added to potassium carbonate (10%) on ice and extracted with ethyl acetate. The organic layer was separated, washed with a solution of saturated sodium chloride, dried (MgSO4), filtered, and evaporated giving 0.086 g (100%) of compound 27, melting point 96° C., enantiomeric excess 88%.
d) Preparation of (S)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1H)-quinolin-2-one (Compound 28)

Compound 27 (0.00038 mol) in hydrochloric acid 3N (9.25 ml) and THF (9.25 ml), was stirred at 60° C. for 24 hours and evaporated, giving 0.18 g (100%) of compound 28, melting point 210° C.
EXAMPLE A.2 a) Preparation of N-[(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methylene)]-p-toluenesulfinamide [(S(S)](Compound 29)

Ti(OEt)4 (0.0419 mol) was added to a mixture of (4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methanone (0.0084 mol) and (S)-(+)-p-toluenesulfinamide (0.0084 mol) in DCE (18 ml). The mixture was stirred and refluxed for 7 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgSO4), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/NH4OH 97/3/0.5), yielding 1.15 g of compound 29 (38%).
The compound N-[(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methylene)]-p-toluenesulfinamide [(S(R)] can be obtained in an analogues way.
B. Preparation of Final Compounds
EXAMPLE B.1 a) Preparation of (S)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone (Compound 30)

Compound 28 (0.00038 mol) was added to a solution of THF (1.8 ml) and NaOH 10N (1.8 ml). BTEAC (0.0019 mol) and methyliodide (0.00076 mol) were added and the mixture was stirred for 2 hours at room temperature. EtOAc was added. The organic layer was separated, dried (MgSO4), filtered, and evaporated giving 0.149 g (83%) of compound 30, enantiomeric excess 86%.


 Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.
Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.References
- [1]
- “R115777 in Treating Patients With Advanced Solid Tumors”
- “R115777 to Treat Children With Neurofibromatosis Type 1 and Progressive Plexiform Neurofibromas”
- R115777 New Drug Application
Angibaud, P.; Venet, M.; Filliers, W.; Broeckx, R.; Ligny, Y.; Muller, P.;Poncelet, V.; End, D. Eur. J. Org. Chem. 2004, 479.
see………….http://onlinelibrary.wiley.com/doi/10.1002/ejoc.200300538/abstract
(b) Filliers, W.; Broeckx, R.;Angibaud, P. U.S. patent, US7572916, 2009.





































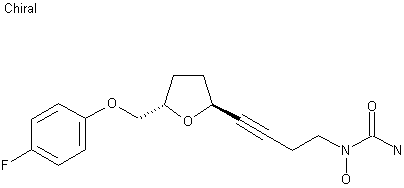
 A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.
A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.
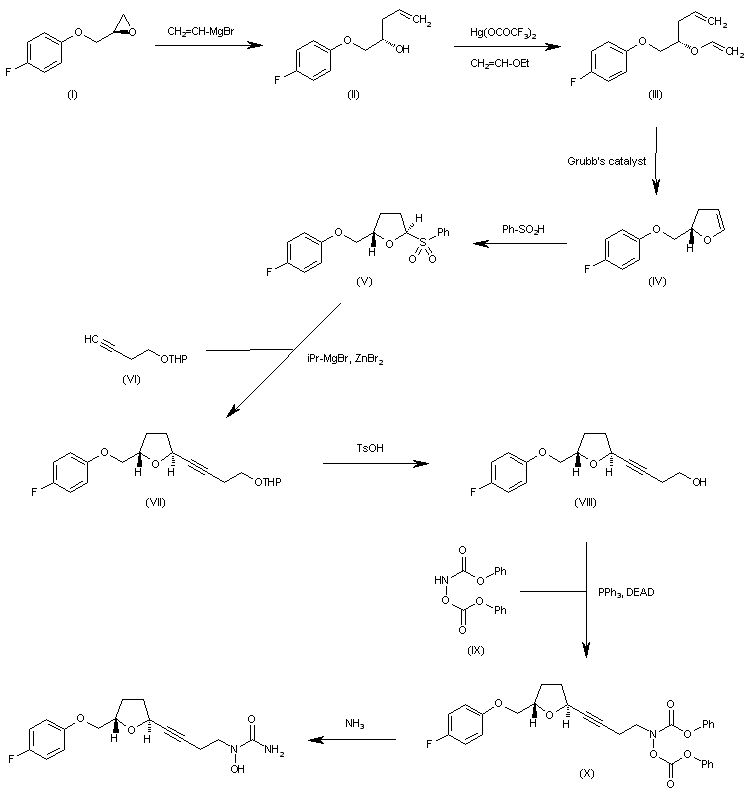 The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.
The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.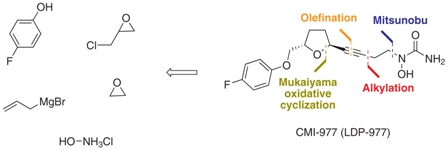





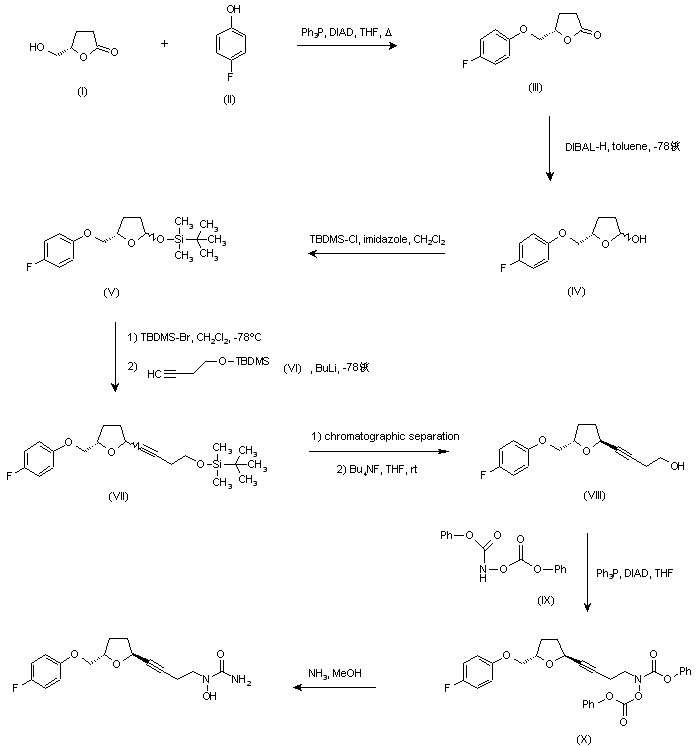 US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.
US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.










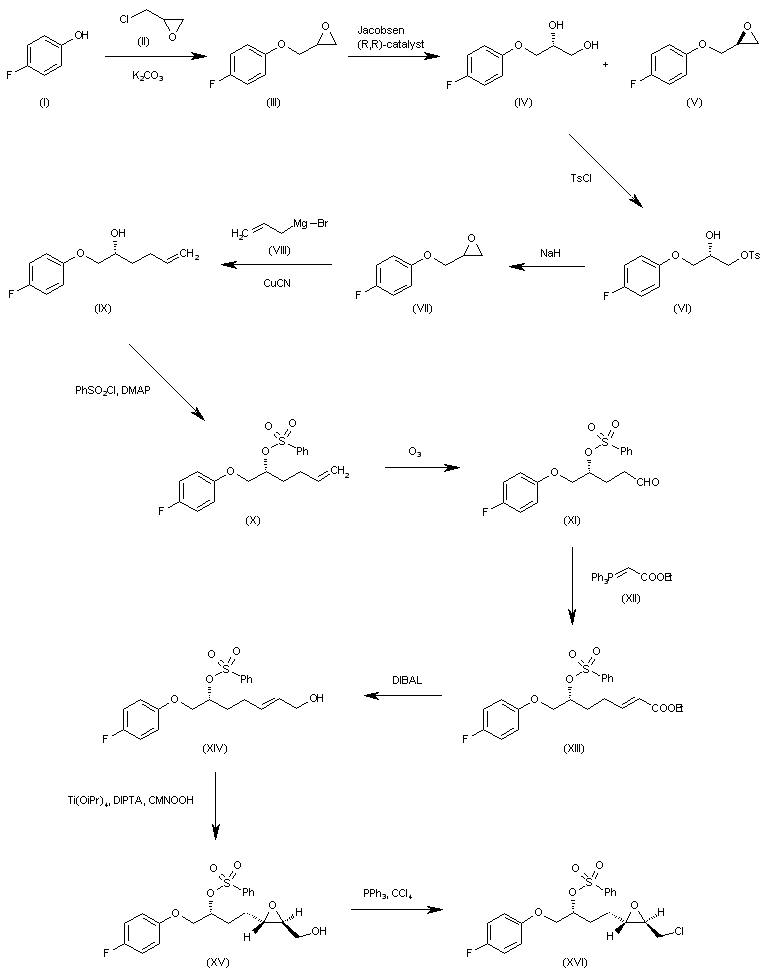 WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….
WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….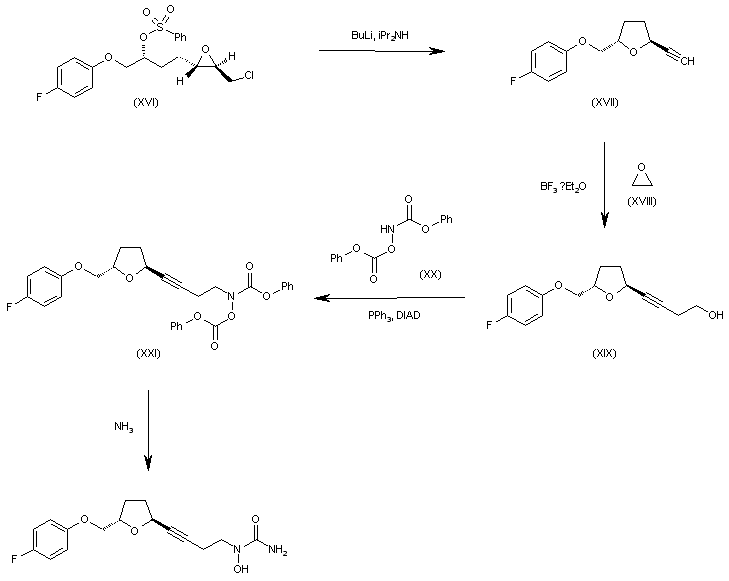 WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.
WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.