












Sun Kim
QbDWorks.com – Quality by Design for Pharma, Biotech, Medical Devices
Dr. Sun K. Kim is a Quality-by-Design Evangelist, transforming how Product Development is executed in the Biologics, Pharmaceutical and Medical Devices industry. In addition, he teaches at Keio University and Stanford University. His current focus of research is Quality-by-Design, Agile Development of Drugs and Therapeutics.
He received his MS and PHD in Mechanical Engineering at Stanford University. Sun was recently a Professor at Keio University in Japan. Prior to Silicon Valley days, he served in the Korean Army and worked at BMW in Munich, Germany
- sun@qbdworks.com
- https://www.linkedin.com/in/kimsunkist
- http://www.leanqbd.com/
- http://qbdworks.com/
- http://www.slideshare.net/kimsunkist
- Twitter…….https://twitter.com/kimsunkist
-
Sun Kim – Google+
https://plus.google.com/104532120968165429422Sun Kim. Worked at QbDWorks. Attended Stanford University. Lives in San Francisco, CA. 2,886 views … QbD Risk Assessment – Quality by Design. 1.
-
Sun Kim – YouTube
https://www.youtube.com/channel/UCoWqD615csn0MUEXDW2O2QgQbDWorks share what works and does not when implementing Quality-by-
Design in the Biotech, Biologics, Pharmaceutical and Medical Devices Industry.
-

Experience

Quality by Design – Founder
QbDWorks
– Present (2 years 6 months)http://QbdWorks.com
Founder of QbDWorks.com

Lecturer
Stanford University
– Present (10 years)Stanford, CA
Teach Design for Manufacturing, Robust Design, Design of Experiments

Master Black Belt in Quality-by-Design, Lean Six Sigma, Sr. Manager
Bayer HealthCare
– (2 years 7 months)Berkeley, CA
Sr. Manager, Master Black Belt in Quality-by-Design, Design for Lean Six Sigma,
Leading Business Process Management

Design for Excellence Evangelist
Abbott
– (1 year 9 months)
Master Black Belt (Lean Six Sigma), Project Management Professional, Scrum Master
Assistant Professor of Graduate School of Systems Design and Management Assistant
Keio University
– (3 years 1 month)
http://www.sdm.keio.ac.jp/en/faculty/kim_s.html
Lecture and advise graduate-level, professional students on system and product design, design thinking, creative brainstorming methodologies, prototyping, project management and business development. Solicited 15 industry project partners. Generated $35,000/year after developing non-degree curriculum for professionals. Co-Investigator of research projects of $50,000: Indoor Location-based Services Technology for Mobile Devices. Consults manufacturing companies (Hitachi, Toshiba) on growth strategies for Service Business Innovation. Others include developing cost simulation tool of product design based on injection molding, design for manufacturing and healthcare delivery systems. Began as a lecturer in Feb. 2008 to co-develop a project-based design curriculum, Active Learning Program Sequence (ALPS), educating over 100 graduate students every year.
Invited as an Assistant Professor in June, 2009.
Research, Teaching Assistant
Stanford University
– (3 years 10 months)
Lectured, coached and managed over 40 multi-disciplinary teams on Design for Manufacturing projects from Biomedical device (Medtronic, Maquet, St. Jude Medical, etc.) and automotive companies (Toyota, Nissan, GM, etc.). Served as the main research associate of Toshiba Corporation Six-Sigma Consulting Inc., developing systems design and manufacturing programs for Toshiba employees. Innovative projects were mobile personal-assistant IT system and agile transportation infrastructure.
Industry-sponsored Projects
Stanford University
– (4 years 10 months)
Maquet Cardiovascular: Coached and led a 3 member team in redesigning the crimping process of Hemashield Grafts, resulting in cost reduction of $75,000 and operators’ medical costs from injuries.
Satiety (Bariatric Surgery Device for Obesity Treatment) Design for Manufacturing Project: Coached a 3 member team in redesigning the packaging and supply chain for the Toga System, resulting in supply chain efficiency of 50% improvement by applying Lean and Errorproofing (Poka Yoke) Techniques.
Medtronic Vascular: Coached a 4 member team in redesigning the manufacturing line of a stent-graft, resulting in 73% reduction of lead time and increase in reliabilty and performance. Observed over 5 vascular and general surgery cases. http://www.youtube.com/watch?v=RkA2TyCsV0A
St. Jude Medical: Led a 4 member team in developing a 7-year supply chain strategy for new service centers of ICD/pacemaker programmers in Europe, Asia, N. and S. America and Oceania. The recommendation consists of an optimized cost model from net present value analysis and AHP location decision modeling that will save $461 million over 7 years and increase customer service rate compared to the existing service centers.
Nissan Motors: Led a 4 member team to construct a 20 year technology / business roadmap of Nissan Fuel Cell powertrain / vehicles which projects $ 4.8 billion revenue. Created a fuel cell vehicle concept design by applying Design for manufacturing tools including market research, manufacturability, and profitability analysis.
Zimmer Orthopedics: Implemented, with five team members, a FEA (finite element analysis) simulation tool with ABAQUS which assists in the development of treating knee osteoarthritis, based on MRI and gait data. http://www.youtube.com/watch?v=47QOdiauHwE
General Motors: Achieved potential cost reduction of $450 per vehicle and reduced 50kg of car weight by replacing wires with conductive coatings and RFID applications with a team of four members.

Design for Six Sigma Research Fellow
Toshiba
– (3 years 7 months)
Developed Design for Six Sigma Curriculum for Toshiba Corp.
Trained engineers, managers in systems design methodologies.
Coached Six Sigma projects.

Medical Device Design Innovation Program Developer
Johnson & Johnson
– (3 months)
Developed an innovation-incubation program to design/develop next generation product/technology with physicians and multidisciplinary design teams.
Design, Manufacturing Consultant
NeoGuide Systems
– (6 months)
Performed Robust Design, Design of Experiment, Developed manufacturing tooling, testing protocols and automated stations.

Reliability Research Assistant
Stanford Linear Accelerator Center
– (7 months)
Developed a reliability decision analysis tool for the LINAC System which will save $83 million per year on the $8 billion International Linear Collider Project. Enhanced reliability of the Accelerator system up to 20% and had increased throughput by linking Failure Mode and Effect Analysis of the Tuner to the evaluation tool.

Optimization/ Structural Analysis Intern
Samsung Electronics
– (1 month)
Improved impact-worthiness (20%) by optimizing design parameters of cell phone cases after performing structural analysis.

Design, Manufacturing Engineer
BMW
– (8 months)
Applied for 1 patent individually and created 3 design proposals as a team in 3 months. Saved $2,760 per month by implementing knowledge database management system. Resolved 3 process problems during 2 weeks of manufacturing
rotation program in Munich assembly plant with the manufacturing engineers.

US – ROK Army Radiology Tech, Company Leader, Manager
Republic of Korea Army
– (2 years 3 months)
Served 20,000 US Army patients as a Radiology Technician, tasks including diagnostic x-ray imaging, upper GI, etc. Managed 12 multi-national soldiers and a radiology department. Was awarded as “the accident-free company.” Increased the availability of the radiology department, which takes care of 20,000 patients, up to 130% by building a forecast schedule planning system.


Education

Stanford University
Ph.D, Mechanical Engineering
Focus in Systems (Product, Service, Business) Design, Design Thinking, Design for Manufacturing and Six Sigma
Activities and Societies: Design Society, ASME, IEEE, INCOSE, ACM
Stanford University
MS, Mechanical Engineering
Focus in Biomedical Device Design, Reliability Engineering, Operations Research
Activities and Societies: KOSEF Academic Fellow

Publications
A New Project-Based Curriculum of Design Thinking with Systems Engineering Techniques
International Journal of System of Systems Engineering
2013
Agile Project Management for Root Cause Analysis Projects
International Conference on Engineering Design
2013
A New Project-Based Curriculum of Design Thinking with Systems Engineering Techniques
Council of Engineering Systems Universities
2012
Evaluation of Design for Service Innovation Curriculum: Validation Framework and Preliminary Results
nternational Journal of Services Technology and Management
2011
A Validation Regarding Effectiveness of Scenario Graph
ASME International Design Engineering Technical Conferences
2011
Wants Chain Analysis: Human-centered Method for Analyzing and Designing Social Systems
International Conference on Engineering Design
2011
Scenario-based Amorphous Design (SAD) Framework for a Location-based Services Technology
Mobile Human Computer Interaction
2010
Transforming Seamless Positioning Technology into a Business using a Systems Design Approach—Scenario-based Amorphous Design
IEEE- International Systems Conference
2010
Design for Service Innovation: A Methodology for Designing Service as a Business for Manufacturing Companies
International Journal of Services Technology and Management
2010
Preliminary Validation of Scenario-based Design for Amorphous Systems
International Conference on Systems Engineering
2010
Tools for Project-based Active Learning of Amorphous Systems Design: Scenario Prototyping and Cross Team Peer Evaluation
ASME International Design Engineering Technical Conferences
2009
Active Learning Project Sequence: Capstone Experience for Multi-disciplinary System Design and Management Education
International Conference on Engineering Design
2009
Demystifying Ambiguity in The Design of Amorphous Systems
International Conference on Systems Engineering
2009
Scenario-based Design for Amorphous Systems
ASME International Mechanical Engineering Congress and Exposition
2008
Analysis and Design Methodology for Recognizing Opportunities and Difficulties for Product-based Services
Information Processing Society of Japan (IPSJ) Journal
2007
Scenario Graph: Discovering new business opportunities and Failure Modes,”
ASME International Design Engineering Technical Conferences
2007
Analysis and Design Methodology for Product-based Services
Annual Conference of the Japanese Society for Artificial Intelligence
2007
Analysis and Design Methodology for Recognizing Opportunities and Difficulties for Product-based Services
PICMET
2007



About QbDWorks…http://qbdworks.com/about/
Are you a Scientist in the Pharmaceutical, Biopharmaceutical or Medical Devices industries?
Then you are probably asking:
- Does Quality-by-Design actually work?
- Or is it just another program like Lean or Six Sigma?
- How do I implement QbD successfully?
- How do I persuade my management?
- What is the first step?
As a QbD practitioner, I had the same questions and am trying to answer them as I test different elements of QbD.
Through our members’ successes and failures in QbD, you can save time by not having to repeat them yourself. There are many lessons learned and knowledge that you can share with your QbD team.
Who are You?

My name is Sun Kim. I currently practice Quality-by-Design, transforming how Product Development is executed in Biopharmaceutical, Pharmaceutical, Biologics, and Medical Device industries.
In addition, I teach at Stanford University. My focus of research is Lean Quality by Design.
I received my MS and PHD in Mechanical Engineering at Stanford University and was recently an Asst. Professor at Keio University in Japan. Prior to Silicon Valley days, I served in the Korean Army and worked at BMW AG in Munich, Germany, where my lifelong pursuit of “Product Development Methodology” began.
Why is an Engineer working in the Bio/pharmaceutical Industry?
Thanks for asking! When working at BMW, as a member of the elite “KREATIV” (Creative) team, my goal was to develop the best technologies and products for the automotive industry. However our approach was somewhat adhoc and heuristics-based. I knew there was a better way.
So I set my heart to learn how best products are developed across all industries. This led me to my PhD research at Stanford University.
Little did I know this would turn out to be more than just a graduate program. On top of the typical coursework, research and publishing, my schedule was packed with hands-on consulting/research projects with GE Healthcare, GE Aviations, Toyota, Nissan, Toshiba, Medtronic, Johnson & Johnson, Startup’s etc.
After contributing to the product development approaches for the Academia, Fortune 500 and Startup’s, I knew my heart was always with Health Care. So I returned to the benches and trenches.
For the last 10 years, I have been working in the Biopharmaceutical, Pharmaceutical and Medical Devices Industry, to make Quality by Design a reality.
So please join me in this Quality by Design journey.
Sign up to connect to the community and receive updates.
Together, we can change how drugs and therapies are developed for our patients and families.
Let’s Connect!
If you’d like to connect, please invite me:
www.linkedin.com/in/kimsunkist/
Previously I worked (full time or consulting) with:

The views expressed on this website are personal opinions and in no way reflect the position of any organization.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus 
 amcrasto@gmail.com
amcrasto@gmail.com






DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus















 GOLDEN TEMPLE
GOLDEN TEMPLE
 .
.





 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar








 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone

%27,_oil_on_canvas_painting_by_Charles_W._Bartlett,_1920,_Honolulu_Academy_of_Arts.jpg)
 tandoori chicken
tandoori chicken





































 .
.
 .
.

































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
 .
.

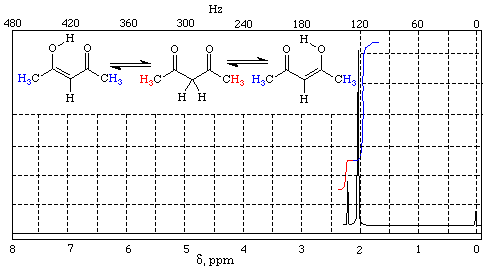

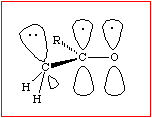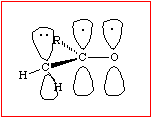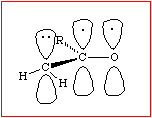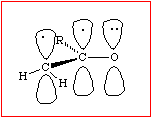






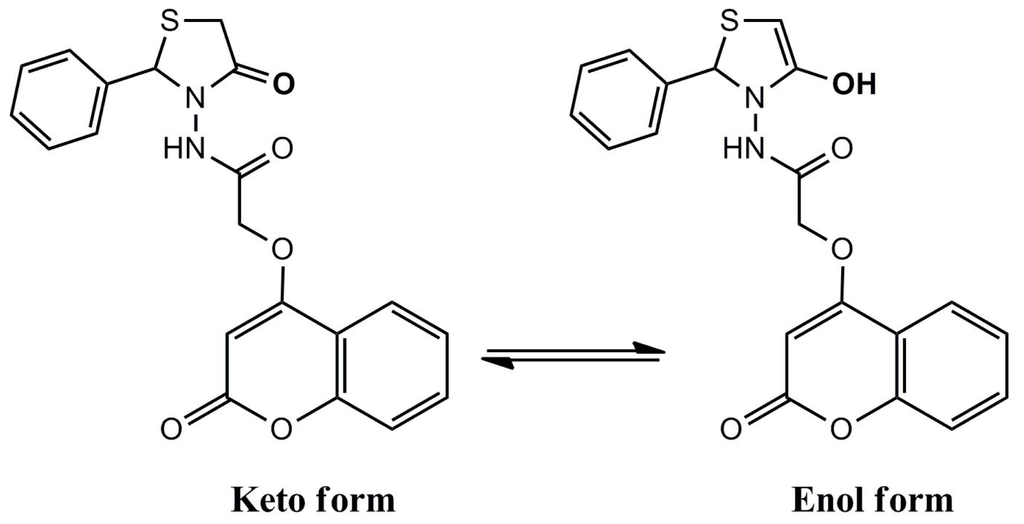

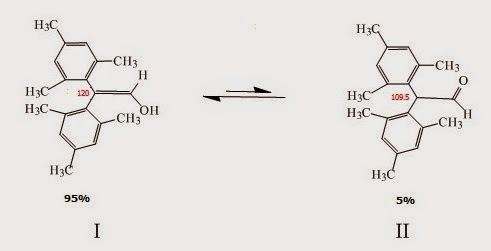
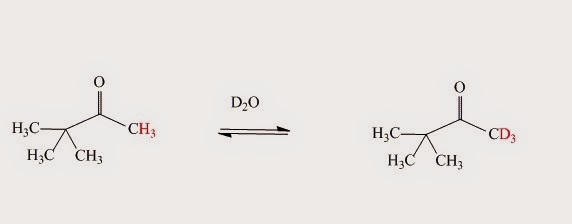


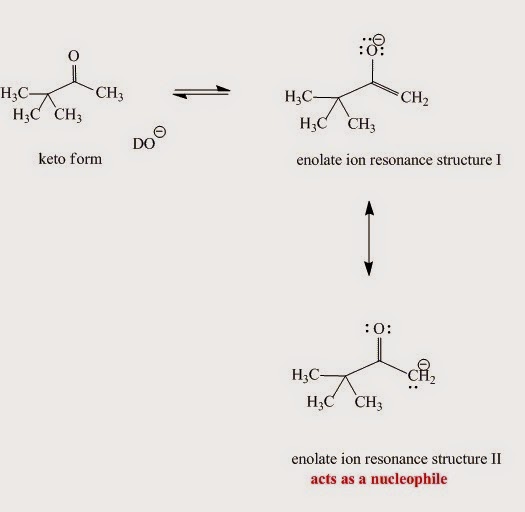


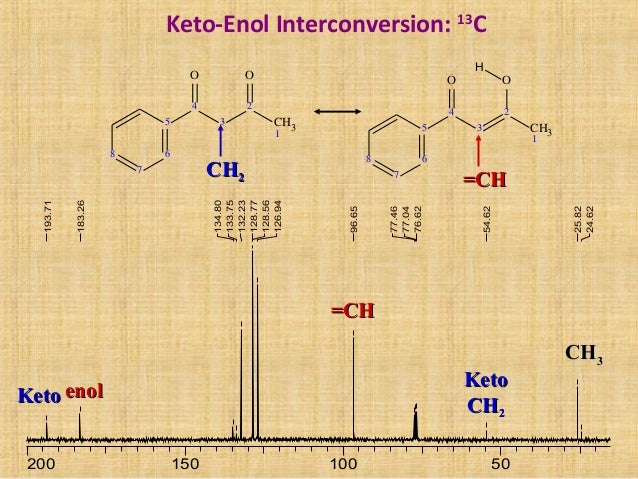 .
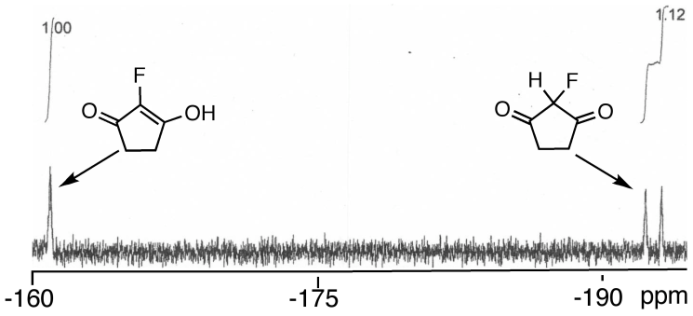
.

 .
.
 ISKCON
ISKCON



.jpg)



































 .
.




















 Winners of the 2014 Elsevier Foundation Awards for Early Career Women Scientists in Developing Countries:
Winners of the 2014 Elsevier Foundation Awards for Early Career Women Scientists in Developing Countries: 










 .
. .
.
 .
. .
. .
. .
. .
.
 .
. .
.




 10000 devout Hindus were present for the Hindu Dharmajagruti Sabha at Amalner, Maharashtra
10000 devout Hindus were present for the Hindu Dharmajagruti Sabha at Amalner, Maharashtra












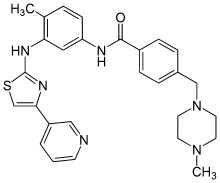












![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_1h.png)
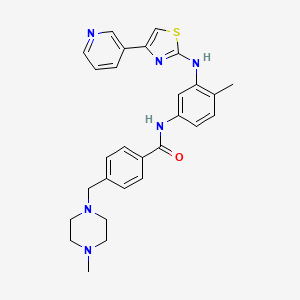
![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_13c.png)












