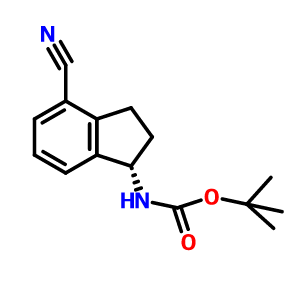
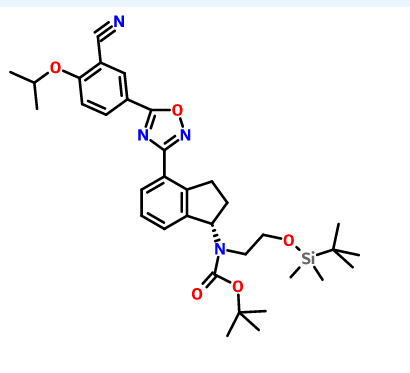
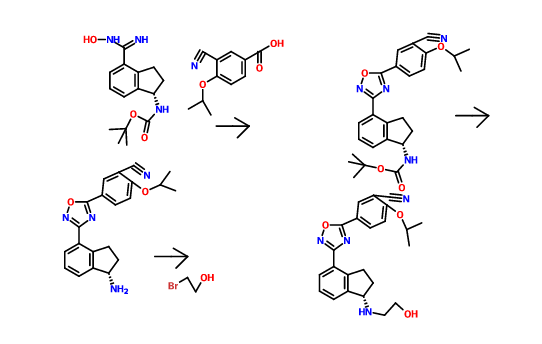
Gamendazole
(E) 3-(1-(2,4-Dichlorobenzyl)-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic Acid
trans-3-(1-Benzyl-6
(E)-3-[1-[(2,4-Dich
- C18H11Cl2F3N2O2
- mw415.193
- RC-MC-110
Heat Shock Protein 90 (HSP90) Inhibitors
University of Kansas Innovator
Gamendazole is a novel drug candidate for male contraception. It is an indazole carboxylic acid derived from lonidamine (LND). Gamendazole produced 100% antispermatogenic effects at 25 mg/kg i.p. in rats, whereas 200 mg/kg was fatal for 60% of rats tested. Since gamendazole produced 100% efficacy, it was tested orally. At a dose of 6 mg/kg, 100% of rats were infertile 4 weeks after a single administration. Complete infertility was maintained for 2 weeks, followed by complete recovery in 4 of 7 rats. The other 3 never recovered fertility. Upon dosing 6 mg/kg orally for 7 days, it produced similar infertility results, but only 2 of 7 rats recovered fertility. There were no abnormalities in rates of conception or abnormal conception in rats who recovered fertility.
Pathology reports were conducted on gamendazole treated rats. At 25 mg/kg i.p., 6 mg/kg oral, and in animals that survived 200 mg/kg i.p., there were no remarkable findings, with no evidence of inflammation, necrosis, tumors, or hemorrhage. There was also a lack of observable behavioral effects at 25 mg/kg i.p., 6 mg/kg oral, and in animals that survived 200 mg/kg i.p. Gamendazole treatment had no effect on testosterone levels, and was reported to affect Sertoli cell function, leading to decreased levels of inhibin B. Low levels of inhibin B were correlated to the infertility of the rat
Female oral contraceptive drugs are widely available in the market by several trade names, including Altravera, Brevicon, Levora, and i-pill, whereas potentially safer, more convenient, and more effective oral male contraceptives are not yet commercially available. However, there are some experimental drugs.AF-2785 1, gamendazole 2, lonidamine 3, and adjudin 4 are most promising among the experimental


Gamendazole was recently identified as an orally active antispermatogenic compound with antifertility effects. The cellular mechanism(s) through which these effects occur and the molecular target(s) of gamendazole action are currently unknown. Gamendazole was recently designed as a potent orally active antispermatogenic male contraceptive agent. Here, we report the identification of binding targets and propose a testable mechanism of action for this antispermatogenic agent. Both HSP90AB1 (previously known as HSP90beta [heat shock 90-kDa protein 1, beta]) and EEF1A1 (previously known as eEF1A [eukaryotic translation elongation factor 1 alpha 1]) were identified as binding targets by biotinylated gamendazole (BT-GMZ) affinity purification from testis, Sertoli cells, and ID8 ovarian cancer cells; identification was confirmed by matrix-assisted laser desorption/ionization-time of flight mass spectrometry and Western blot analysis. BT-GMZ bound to purified yeast HSP82 (homologue to mammalian HSP90AB1) and EEF1A1, but not to TEF3 or HBS1, and was competed by unlabeled gamendazole. However, gamendazole did not inhibit nucleotide binding by EEF1A1.
Gamendazole binding to purified Saccharomyces cerevisiae HSP82 inhibited luciferase refolding and was not competed by the HSP90 drugs geldanamycin or novobiocin analogue, KU-1. Gamendazole elicited degradation of the HSP90-dependent client proteins AKT1 and ERBB2 and had an antiproliferative effect in MCF-7 cells without inducing HSP90. These data suggest that gamendazole may represent a new class of selective HSP90AB1 and EEF1A1 inhibitors. Testis gene microarray analysis from gamendazole-treated rats showed a marked, rapid increase in three interleukin 1 genes and Nfkbia (NF-kappaB inhibitor alpha) 4 h after oral administration. A spike in II1a transcription was confirmed by RT-PCR in primary Sertoli cells 60 min after exposure to 100 nM gamendazole, demonstrating that Sertoli cells are a target. AKT1, NFKB, and interleukin 1 are known regulators of the Sertoli cell-spermatid junctional complexes. A current model for gamendazole action posits that this pathway links interaction with HSP90AB1 and EEF1A1 to the loss of spermatids and resulting infertility.
Synthesis








…………………….
2-Halo benzoic acid is converted into aroyl chloride and then to aroyl cyanide in an overall yield of 82%. Aroyl cyanides 5 are converted to 2-halophenyl glyoxylate ester 7 via ketoamide 6 in 85% yields as shown in Scheme below. Direct conversion of aroyl cyanide 5 to ester 7 is also reported[ U.S. Patent 4,596,885, 1986 .] but with lesser yields.



Preparation of (E) 3-(1-(2,4-Dichlorobenzyl)-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic Acid (R = CF3) (Gamendazole) (2)
- DOI:
- 10.1080/00397911.2012.696306
Arava Veerareddya*, Gogireddy Surendrareddya & P. K. Dubeyb
pages 2236-2241
Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemistry
Volume 43, Issue 16, 2013
trans 3-[l- (l^-dichlorobenzy^-ό-trifluoromethyl-lH-indazol-S-ylj-acrylic acid (RC-MC-110) is provided.

EXAMPLE 2: Synthesis of a-ll-fl^-dichlorobenzyn-ό-trifluoromethyl-lH-indazol-S-yll-acrylic acid (RC-MC-110)
Step 1 : 2-(2-nitro-4-trifluoromethylphenyl)-malonic acid dimethyl ester.
Dimethyl malonate (59.7 g, 0.44 mol) was added dropwise to a stirred solution of potassium tert-butoxide (51 g, 0.44 mol) in dry t-butanol (500 mL). To the resultant suspension, a warm solution of 2-chloro-5-trifluoromethylnitrobenzene (50 g, 0.22 mol) in t-butanol (100 mL) was added and the mixture was refluxed for 6 h (reaction monitored by TLC). After completion of the reaction, most of the t-butanol was distilled off under vacuum, and chilled water was then added to the reaction mixture. The pH was adjusted to neutral with dilute hydrochloric acid, which resulted in the precipitation of the product. The mixture was stirred for 30 minutes and the product was filtered off (68 g, 95%). This material was used without further purification in the next step. A small amount was crystallized (EtOAc/hexane, 4:6) for analysis, to yield a yellow crystalline material, mp 65-67 0C. 1H NMR (CDCl3) 8.30 (s, 1 H), 7.92 (d, J = 8.4 Hz, 1 H), 7.69 (d, J = 8.4 Hz, 1 H), 5.37 (s, 1 H), 3.80 (s, 6 H). MS (FAB) m/z: 322.1 (M+ + 1).
Step 2: (2-nitro-4-trifluoromethylphenyl)-acetic acid methyl ester.
2-(2-Nitro-4-trifluoromethylphenyl)-malonic acid dimethyl ester (68 g, 0.21 mol) was dissolved in dimethyl sulfoxide (200 mL). Sodium chloride (34 g, 0.58 mol) and water (60 mL) were added and the mixture was stirred for 16-20 h at 120 0C (reaction monitored by TLC). The reaction mixture was then cooled to room temperature and quenched into water, which caused precipitation of the product. After stirring for 30 minutes, the product (45 g, 80%) was isolated by filtration. The product was used without further purification in the next reaction. A small sample was crystallized (EtOAc/hexane, 2:8) for analysis, to yield yellow crystals, mp 104-105 0C. 1H
NMR (CDCl3) 8.3 (s, 1 H), 7.88 (d, J = 8.4 Hz, 1 H), 7.50 (d, J = 8.4 Hz, 1 H), 4.12 (s, 2 H), 3.60 (s, 3 H). MS (FAB) m/z: 275.2 (M+ + 1).
Step 3: (2-Acetylamino-4-trifluoromethylphenyl)-acetic acid methyl ester.
Hydrogenation and acetylation of (2-nitro-4-trifluoromethylphenyl)-acetic acid methyl ester (25 g, 0.095 mol) in the presence of 5% Pd-C (2.5 g, 50% wet) and acetic anhydride (38 g, 0.37 mol) in toluene (200 mL) was carried out under vigorous stirring at room temperature and atmospheric pressure for about 4-5 h (reaction monitored by TLC). The catalyst was removed by filtration and washed with toluene two times. The combined organics were evaporated in vacuo to yield the product (24.8 g, 95%), which was used without further purification in the next step. A small sample was crystallized from hexane to yield the product as a yellow solid, mp 92-94 0C. H NMR (CDCl3) 8.86 (s, 1 H), 8.21 (s, 1 H), 7.36 (d, J = 8.1 Hz, 1 H), 7.31 (d, J = 8.1 Hz, 1 H), 3.74 (s, 3 H), 3.68 (s, 2 H), 2.23 (s, 3 H). Step 4: ό-Trifluoromethyl-lH-indazole^-carboxylic acid methyl ester.
To a solution of (2-acetylamino-4-trifluoromethylphenyl)-acetic acid methyl ester (16 g, 0.058 mol) in acetic acid (50 mL) was added dropwise t-butyl nitrite (90%) (7.35 g, 0.063 mol) over a period of 20 min. at 90-95 0C. The mixture was then stirred for 0.5 h at 95 0C, poured into cold water and stirred for 1 h. The precipitates were collected by filtration and washed with water. The crude material was dissolved in ethyl acetate and dried over sodium sulfate. The solvent was removed in vacuo. This material (13.4 g, 95%) was used without further purification in the next step. A small sample was crystallized from ethyl acetate to yield a white solid, mp 240-242 0C. H NMR (DMSO-d-6) 8.25 (d, J = 8.5 Hz, 1 H), 8.04 (s, 1 H), 7.58 (d, J = 8.5 Hz, 1 H), 3.95 (s, 3 H). MS (FAB) m/z: 245.1 (M+ + 1).
Step 5: l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester.
ό-Trifluoromethyl-lH-indazole-S-carboxylic acid methyl ester (2.75 g, 0.0112 mol) was dissolved in acetonitrile (50 mL), and potassium carbonate (1O g, 0.07 mol), 2,4-dichlorobenzyl chloride (2.42 g, 0.01239 mol) and tetrabutylammonium iodide (catalytic) were added. The reaction mixture was heated to reflux and refluxed for 2 h under good stirring. The progress of the reaction was monitored by TLC. After completion of the reaction, potassium carbonate was filtered while hot and then washed with acetone. The combined solvents were distilled off under reduced pressure to afford the crude mixture of Nl and N2 benzylated products. The isomers were separated by column chromatography (silica gel, eluent started with hexane then changed to 8:2 hexane, ethyl acetate). l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester. Yield: 3.62 g (80%), white crystals mp 118-120 0C. ‘ H NMR (CDCl3) 8.39 (d, J = 8.4 Hz, 1 H) 7.74 (s, 1 H), 7.57 (d, J = 8.4 Hz, 1 H), 7.45 (d, J = 2.1 Hz, 1 H), 7.12 (dd, J = 8.4 and 2.1 Hz, 1 H), 6.78 (d, J = 8.4 Hz, 1 H), 5.82 (s, 2 H), 4.07 (s, 3 H). MS (FAB) m/z: 403 (M+ + 1).Z-^^-DichlorobenzylJ-δ-trifluoromethyl-ZH-indazole-S-carboxylic acid methyl ester. Yield: 680 mg (15%), white crystals mp 132-134 0C. ‘ H NMR (DMSO-d-6) 8.27 (s, 1 H), 8.20 (d, J = 8.7 Hz, 1 H), 7.76 (d, J = 1.8 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.30 (dd, J = 8.3 and 1.8 Hz, 1 H), 6.78 (d, J = 8.3 Hz, 1 H), 6.17 (s, 2 H), 3.96 (s, 3 H).
Step 6: [l-(2.4-Difluorobenzyl)-6-trifluoromethyl-lH-indazol-3-yl1-methanol.
l -(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester (3.0 g, 0.0075 mol) dissolved in CH2Cl2(50 mL) was cooled to -78 0C. DIBAL-H (8.18 mL, 0.00818 mol) was added slowly dropwise via a syringe under an argon blanket over a period of 15 minutes. After the complete addition of DIBAL-H, the reaction mixture was stirred at -78°C for another 2 h (reaction monitored by TLC). The reaction was quenched carefully with methanol at -78 0C. The reaction mixture was then carefully poured into water and the layers were separated. The organic layer was washed with water and dried over sodium sulfate. Removal of the solvent yielded the crude alcohol (2.6 g, 93%), which was used without purification in the next step. The alcohol was a white solid, mp 137-139 0C. 1H NMR (CDCl3) 7.97 (d, J = 8.4 Hz, 1 H), 7.66 (s, 1 H), 7.44 (d, J = 2.0 Hz, 1 H), 7.42 (d, J = 8.5 Hz, 1 H), 7.12 (dd, J = 8.3 and 2.0 Hz, 1 H), 6.93 (d, J = 8.3 Hz, 1 H), 5.65 (s, 2 H), 5.09 (s, 2 H). MS (FAB) m/z: 375 (M+ + 1).Step 7: l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carbaldehvde.
[l-(2,4-Difluorobenzyl)-6-trifluoromethyl-lH-indazol-3-yl]-methanol (3.75 g, 0.01 mol) was dissolved in CH2Cl2 (100 mL) and manganese(IV)oxide (8.7 g, 0.1 mol) was added and stirred for 2-3 h at room temperature (reaction monitored by TLC). The solids were removed by filtration and the removal of the CH2Cl2 in vacuo yielded the crude aldehyde. The aldehyde was used without further purification in the next step. The aldehyde (3.54 g, 95%) was a white solid, mp 97-98 0C. 1H NMR (CDCl3) 10.25 (s, 1 H), 8.45 (d, J = 8.5 Hz, 1 H), 7.79 (s, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.48 (d, J = 2.0 Hz, 1 H), 7.20 (dd, J = 8.3 Hz and 2.0 Hz, 1 H), 6.93 (d, J = 8.3 Hz, 1 H), 5.79 (s, 2 H). MS (FAB) m/z: 373 (M+ + 1).
Step 8: 3-ri-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazol-3-yll-acrylic acid ethyl ester.
l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carbaldehyde (2.0 g, 0.00536 mol) was dissolved in CH2Cl2 (50 niL) and Wittig reagent (carbethoxymethylene) triphenylphosphorane (1.06 g, 0.0536 mol) was added to the solution. The homogeneous reaction mixture was heated to reflux in an oil bath for 12 h. The reaction progress was monitored by TLC. The reaction mixture was cooled to room temperature and worked up by quenching into water and separating the organic layer. Removal of the CH2Cl2 yielded the crude product, which was purified by column chromatography to yield the pure product (2.25 g, 95%) as a white solid, mp 186-188 0C. 1H NMR (CDCl3) 8.08 (d, J = 8.5 Hz, 1 H), 7.99 (d, J = 16.2 Hz, 1 H), 7.74 (s, 1 H), 7.52 (d, J = 8.5 Hz, 1 H), 7.47 (d, J = 2.0 Hz, 1 H), 7.16 (dd, J = 8.3 and 2.0 Hz, 1 H), 6.84 (d, J = 8.3 Hz, 1 H), 6.82 (d, J = 16.2 Hz, 1 H), 5.72 (s, 2 H), 4.32 (q, J = 7.1 Hz, 2 H), 1.38 (t, J = 7.1 Hz, 3 H). MS (FAB) m/z: 443 (M+ + 1).It will be appreciated that the acrylic acid ethyl ester can be hydrogenated using 5% Pd-C in the presence of methanol, DCM at RT and 1 atm-pressure to give the propionic acid ester derivative. For example, treatment under such conditions yields 3-[l-(2,4-dichlorobenzyl)-6- trifluoromethyl-lH-indazol-3-yl]-propionic acid ethyl ester (JWS-2-70).
Step 9: l-(2,4-Dichlorobenzyl‘)-3-r6-trifluoromethyl-lΗ-indazol-3-yll-acrvlic acid.

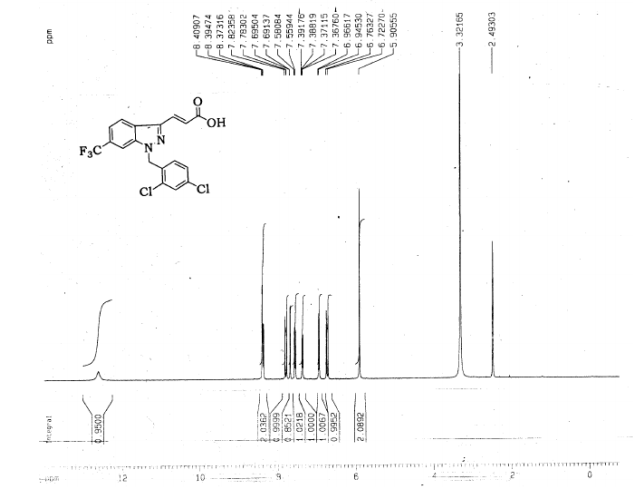
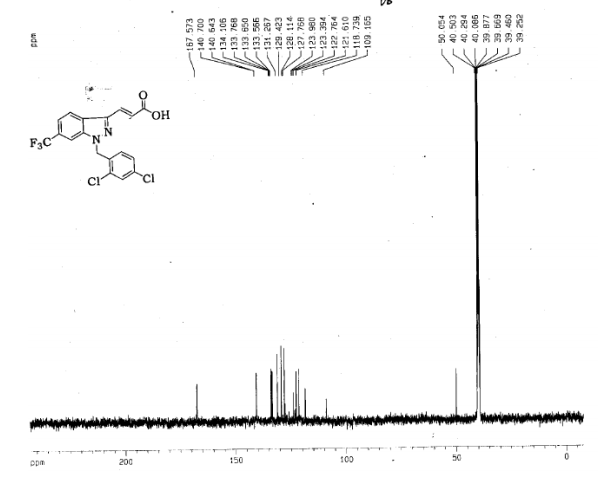
l-(2,4-Dichlorobenzyl)-3-[6-trifluoromethyl-lH-indazol-3-yl]-acrylic acid ethyl ester (2.0 g, 0.0045 mol) was dissolved in a mixture of tetrahydrofuran (50 mL) and methanol (25 mL). A lithium hydroxide solution (0.33 g, 0.013 mol lithium hydroxide in 7.5 mL water) was added slowly at room temperature under good stirring. The reaction mixture was then warmed to 40 0C and held at that temperature for 2 h. The reaction mixture was diluted with water and extracted with ethyl acetate in order to remove neutral impurities. The layers were separated and the aqueous layer was cooled to 0 0C and then acidified with 20% sulfuric acid to pH 2. White solids precipitated and were filtered and dried to constant weight. The crude product was recrystallized from ethyl acetate and hexane (1 :1) to afford the pure product (1.68 g, 90%) as a white solid,
REFERENCES
- 1. Corsi , G. ; Palazzo , G. ; Germani , C. ; Barcellona , P. S. ; Silvestrini , B. 1-Halobenzyl-1H-indazole-3-carboxylic acids: A new class of antispermatogenic agents . J. Med. Chem. 1976 , 19 , 778
- 2. Palazzo , G. ; Corsi , G. ; Baiocchi , L. ; Silvestrini , B. Synthesis and pharmalogical properties of 1-substituted-3-dimethylaminoalkoxy-1H-indazoles . J. Med. Chem. 1966 , 9 , 38 – 41 .
- 3. Silvestrini , B. Basic and applied research in the study of indazole carboxylic acids . Chemotherapy 1981 , 27 ( Suppl.2 ), 9 – 20 .
- 4. Silvestrini , B. ; Palazzo , G. ; De Gregorio , M. D. 3-Lonidamine and related compounds . Progr. Med. Chem. 1985 , 21 , 111 – 135 .
- 5. Cheng , C. Y. ; Silvestrini , B. ; Grima , J. ; Mo , M. Y. ; Zhu , L. J. ; Johnsson , E. ; Saso , L. ; Leone , M. G. ; Palmery , M. ; Mruk , D. Two new male contraceptives exert their effects by depleting germ cells prematurely from the testes . Biol. Reprod. 2001 , 65 , 449 – 461 .
- 6. Xia , W. ; Mruk , D. D. ; Lee , W. M. ; Ceng , C. Y. Unraveling the molecular targets pertinent to junction restructuring events during spermatogenesis using the Adjudin-induced germ cell depletion model . J. Endocrinol. 2007 , 192 , 563 – 583 .
- 7. Cheng , C. Y. ; Mruk , D. D. ; Silvestrini , B. ; Bonanomi , M. ; Wong , C. H. ; Siu , M. K. Y. ; Lee , N. P. Y. ; Mo , M. Y. AF-2364 [1-(2,4-dichlorobenzyl)-1H-indazole-3-carbohydrazide] is a potential male contraceptive: A review of recent data . Contraception2005 , 72 , 251 – 261 .
- 8. Tash , J. S. ; Attardi , B. ; Hild , S. A. ; Chakrasali , R. ; Jakkarg , S. R. ; Georg , G. I. A novel potent indazole carboxylic acid derivative blocks spermatogenesis and is contraceptive in rats after a single oral dose . Biol. Reprod. 2008 , 78 , 1127 – 1138 .
- 9. Sarkar , O. ; Mathur , P. P. Adjudin-mediated germ cell depletion alters the anti-oxidant status of adult rat testes . Mol. Reprod. Dev. 2009 , 76 , 31 – 37 .
- 10. Mok , K.-W. ; Mruk , D. D. ; Lie , P. P. Y. ; Lui , W.-Y. ; Cheng , C. Y. Adjudin, a potential male contraceptive, exerts its effects locally in the seminiferous epithelium of mammalian testes. Reproduction. 2011, 141, 571–580.
- 11. Wang , H. ; Chen , X. X. ; Wang , L.-R. ; Mao , Y.-D. ; Zhou , Z. M. ; Sha , J.-H. AF-2364 is a prospective spermicide candidate .Asian J. Androl. 2010 , 12 , 322 – 335 .
-
- “Gamendazole”. NextBio. www.nextbio.com. Retrieved 31 July 2011.
- Tash, Joseph (July 2008). “A Novel Potent Indazole Carboxylic Acid Derivative Blocks Spermatogenesis and Is Contraceptive in Rats after a Single Oral Dose”. Biology of Reproduction 78 (6): 1127–1138. doi:10.1095/biolreprod.106.057810. PMID 18218612.
Chakrasali, R.; Jakkaraj, S.R.; Tash, J.S.; Hild, S.A.; Attardi, B.; Georg, G.I.
Design, synthesis and in vivo evaluation of Gamendazole(R), a novel orally active male contraceptive agent
228th Am Chem Soc (ACS) Natl Meet (August 22-26, Philadelphia) 2004, Abst MEDI 305
| CHENG C.Y. ET AL: “Two New Male Contraceptives Exert Their Effects by Depleting Germ Cells Prematurely from the Testis” BIOLOGY OF REPRODUCTION, SOCIETY FOR THE STUDY OF REPRODUCTION, CHAMPAIGN, IL, US, vol. 65, no. 2, 1 August 2001 (2001-08-01), pages 449-461, XP002547492 ISSN: 0006-3363 | ||
| 2 | * | GATTA F. ET AL: “Pyrazolo[3,4-d]pyrimidines. Related to Lonidamine” JOURNAL OF HETEROCYCLIC CHEMISTRY, HETEROCORPORATION. PROVO, US, vol. 26, no. 3, 1 March 1989 (1989-03-01), pages 613-618, XP002547493 ISSN: 0022-152X |
| US3895026 * | Feb 9, 1973 | Jul 15, 1975 | Acraf | Substituted 1-benzyl-1h-indazole-3-carboxylic acids and derivatives thereof |
| WO2003097063A1 * | May 5, 2003 | Nov 27, 2003 | Bayer Ag | Derivatives of 2-(1-benzyl-1h-pyrazolo (3, 4-b)pyridine-3yl) -5-(4-pyridinyl)-4-pyrimidine amine and the use thereof as guanylate cyclase stimulators |
| WO2006015263A2 * | Jul 29, 2005 | Feb 9, 2006 | Duan Jian-Xin | Lonidamine analogs |
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
(E)-3-[1-[(2,4-Dichlorophenyl)methyl]-6-(trifluoromethyl)indazol-3-yl]prop-2-enoic acid[1]
|
|
| Other names
trans-3-(1-Benzyl-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic acid)
|
|
| Identifiers | |
| 877773-32-5 |
|
| ChemSpider | 9387234 |
| Jmol-3D images | Image |
| PubChem | 11212172 |
| Properties | |
| C18H11Cl2F3N2O2 | |
| Molar mass | 415.19 g·mol−1 |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 
/////////


 3D
3D
























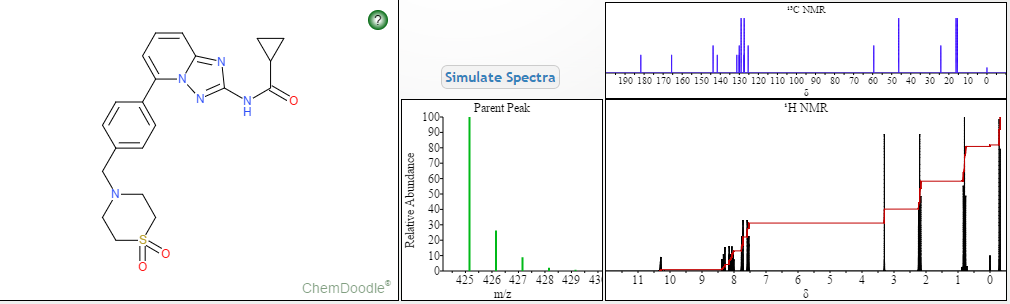
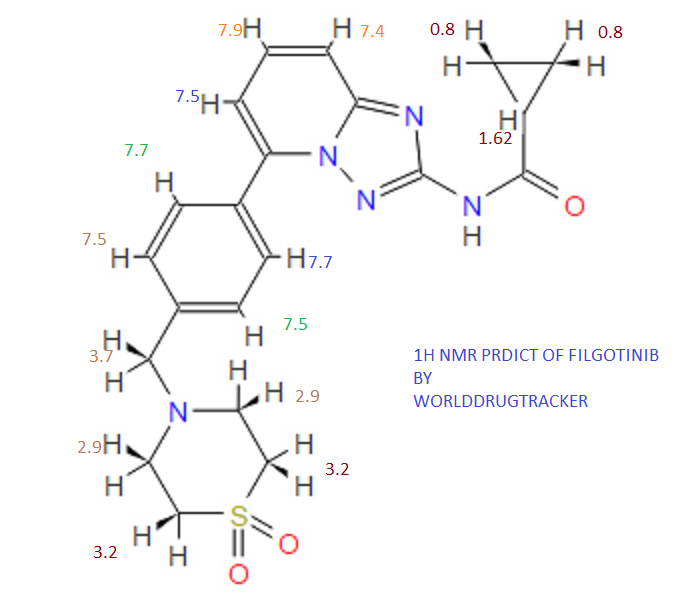
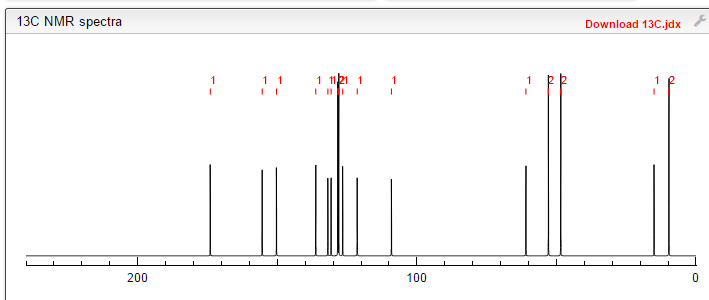
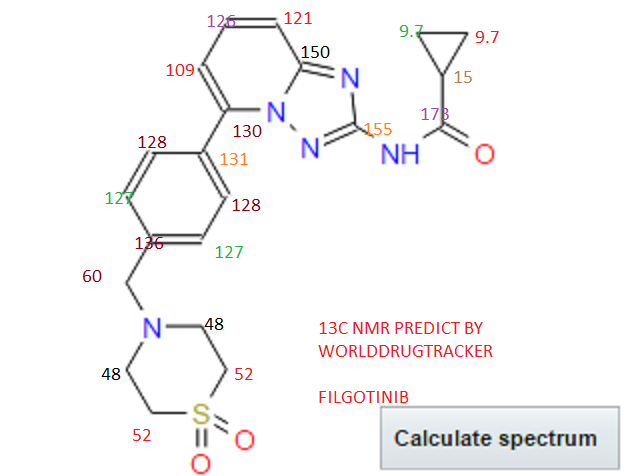
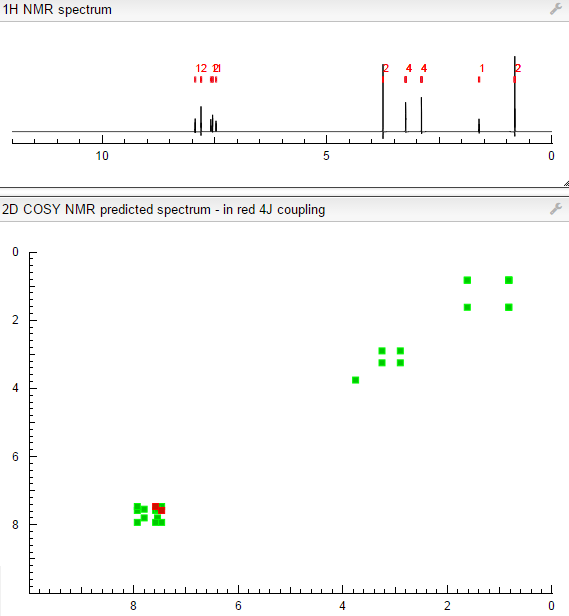


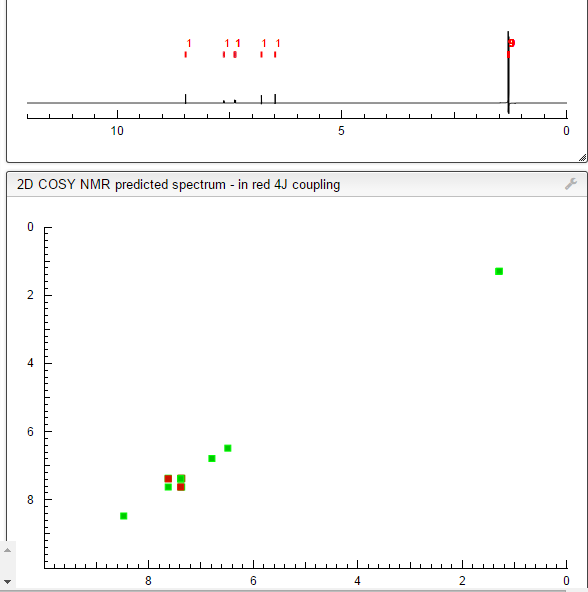
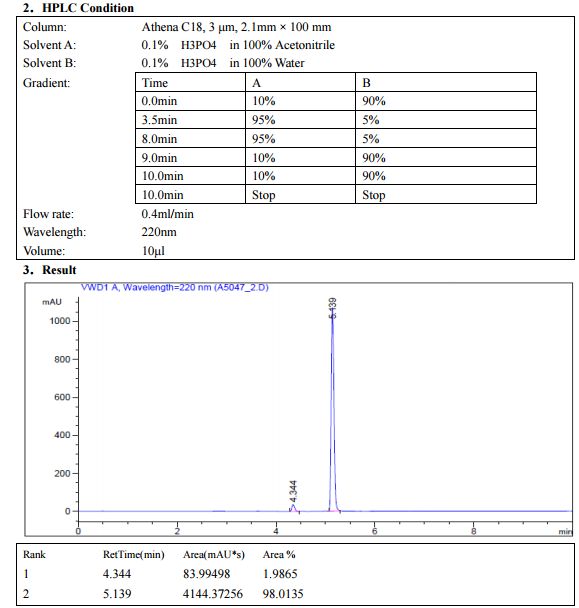

 .
.







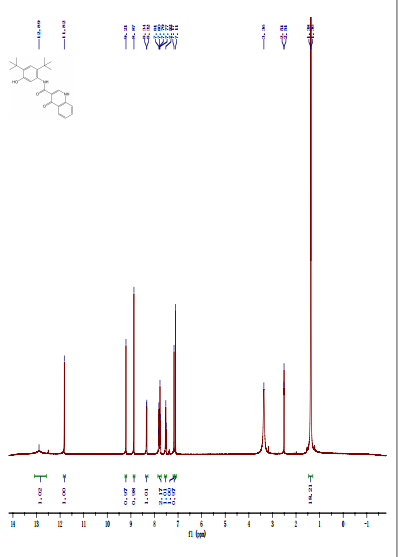
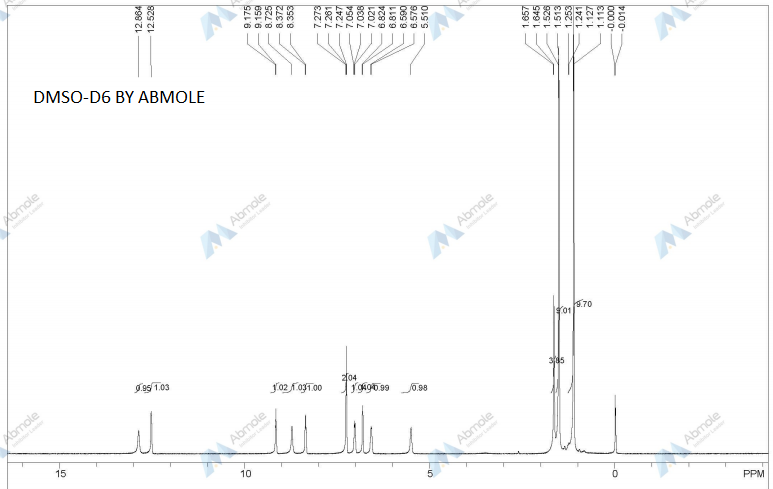
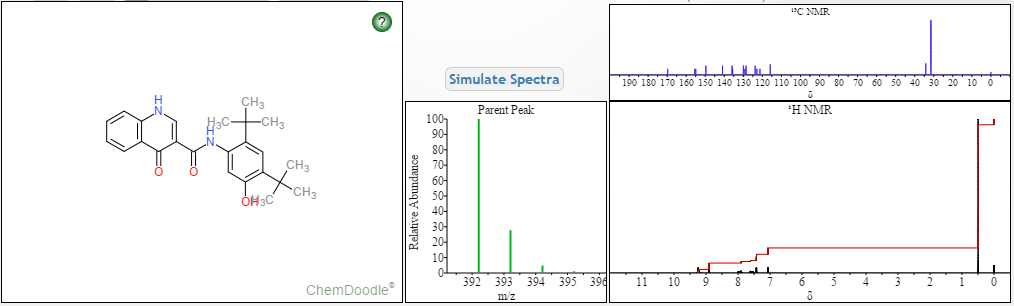
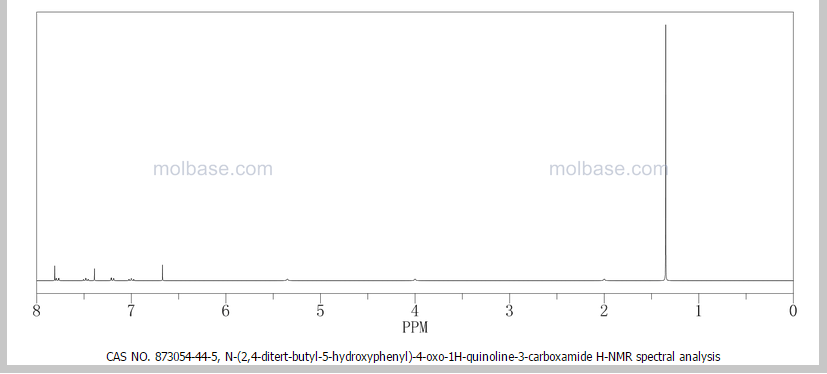
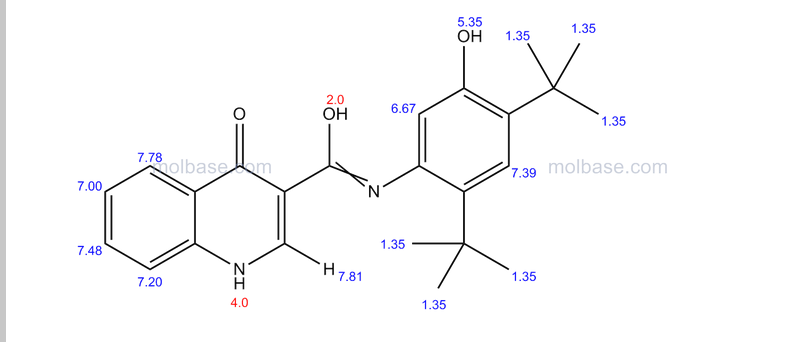
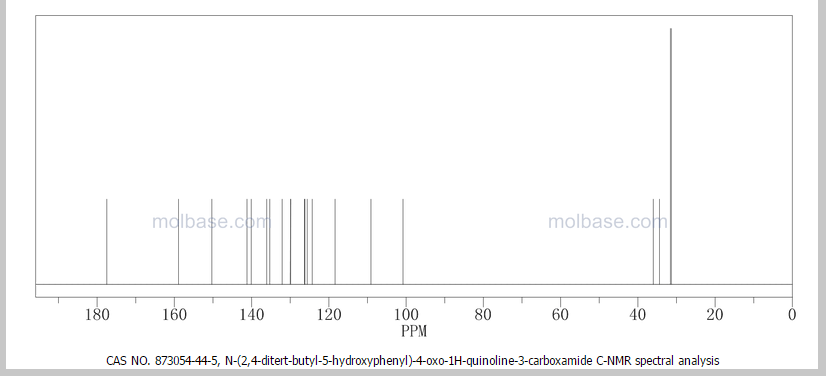
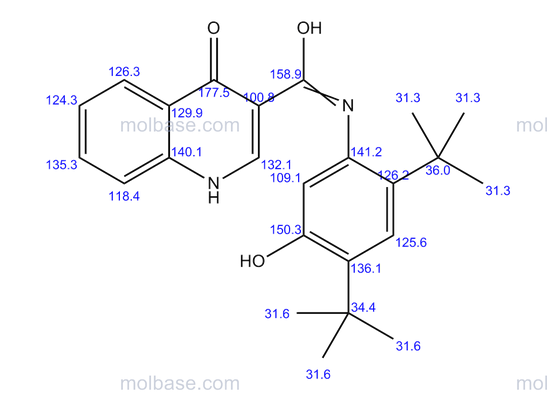



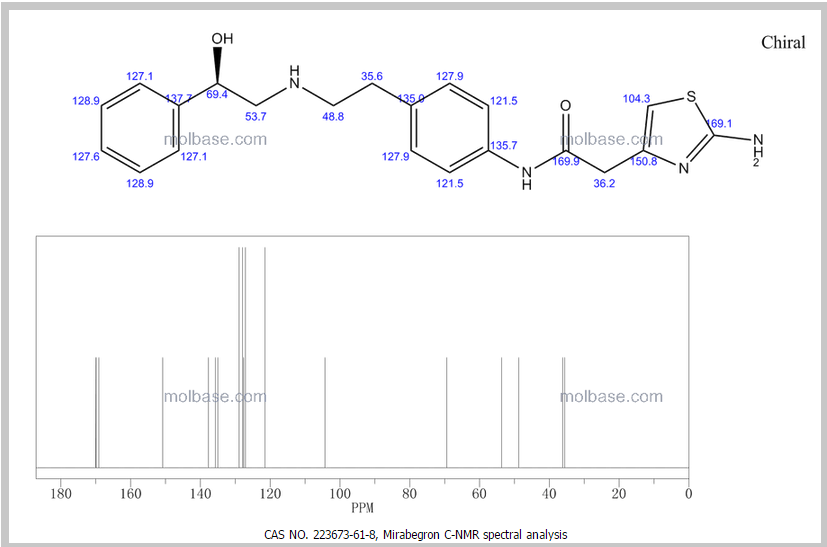
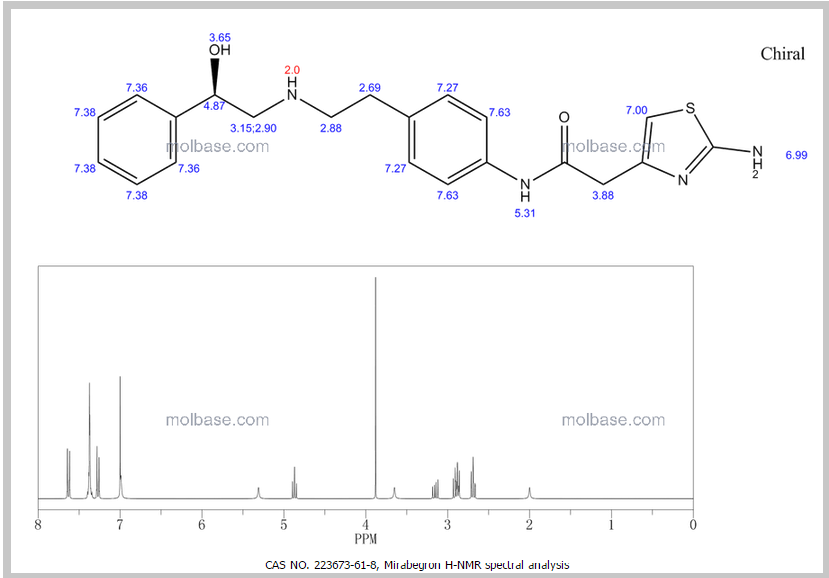
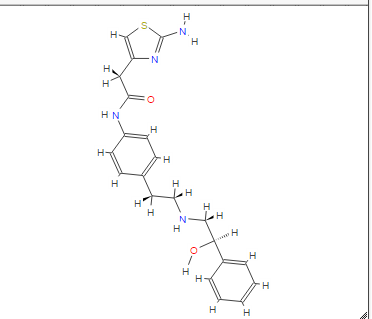
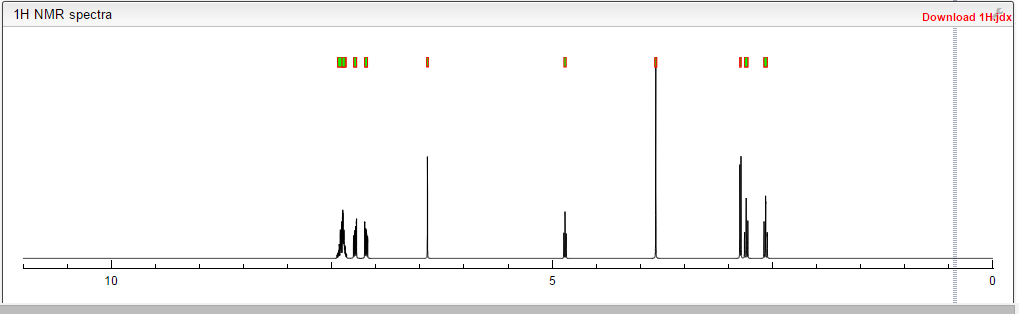
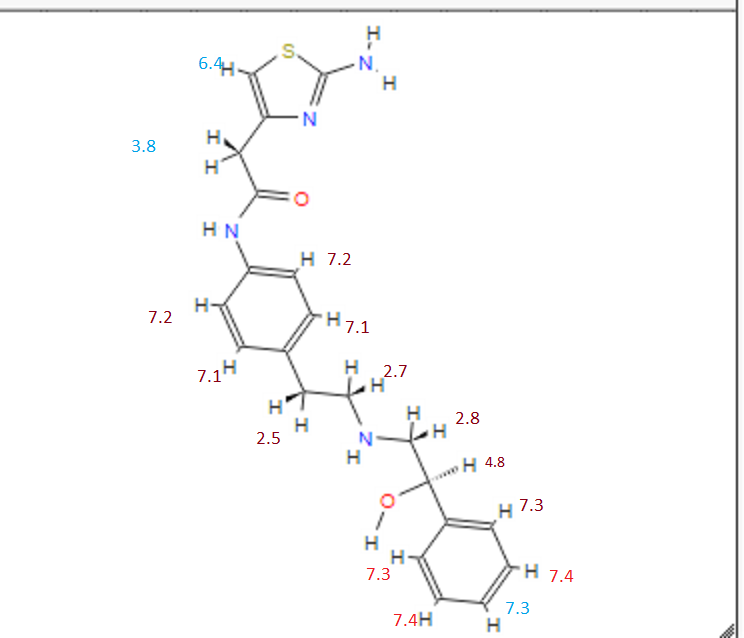
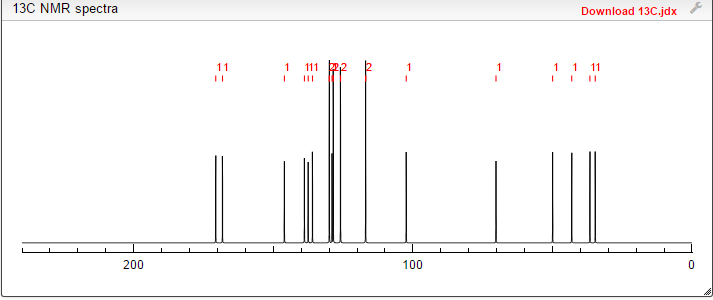
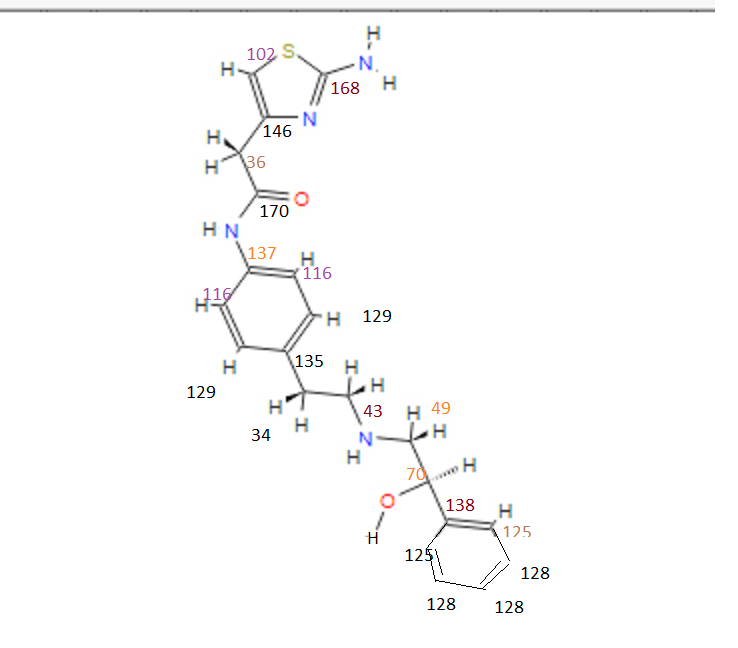
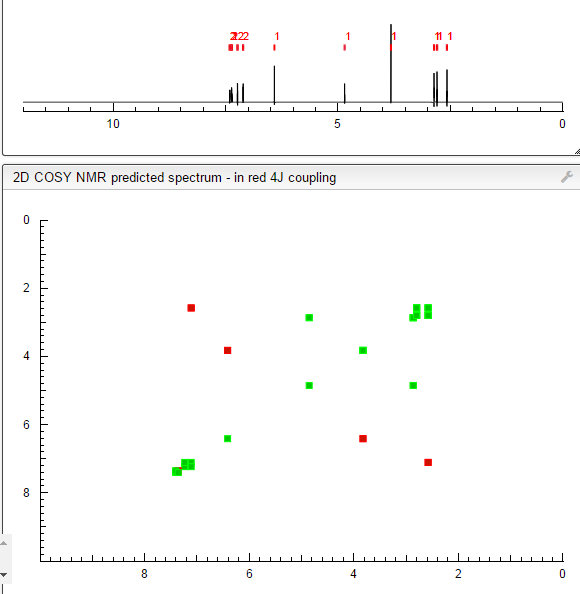































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..























































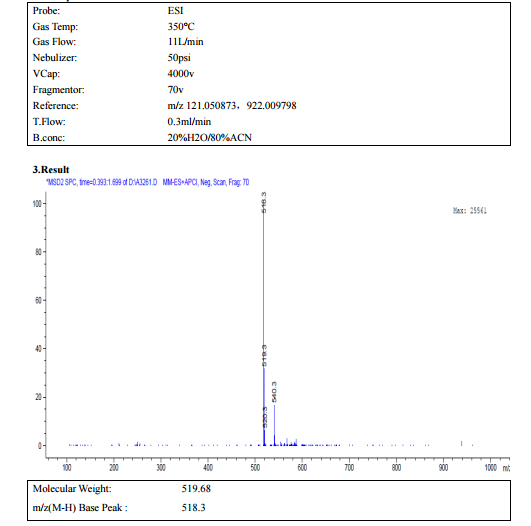

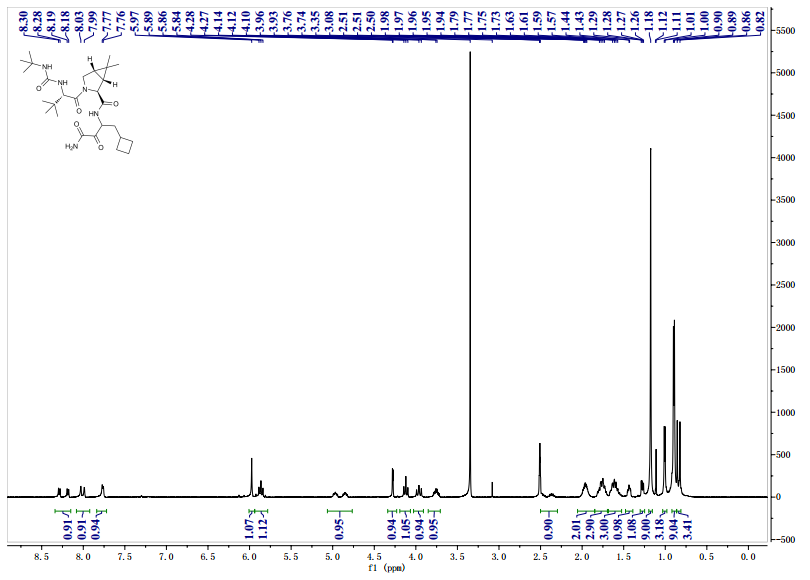


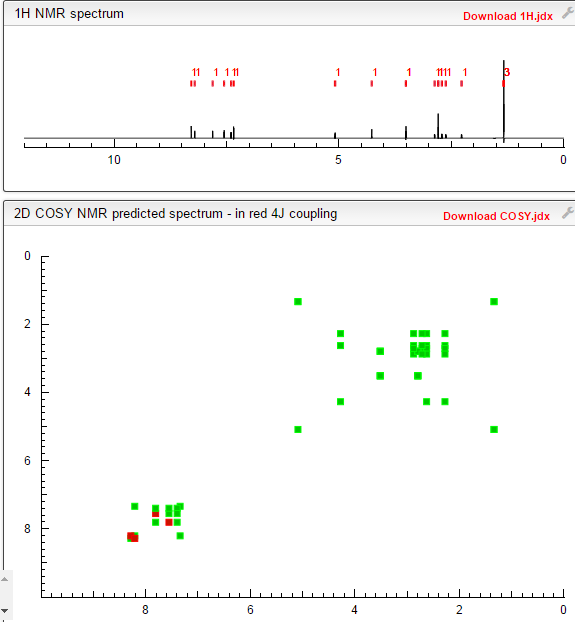
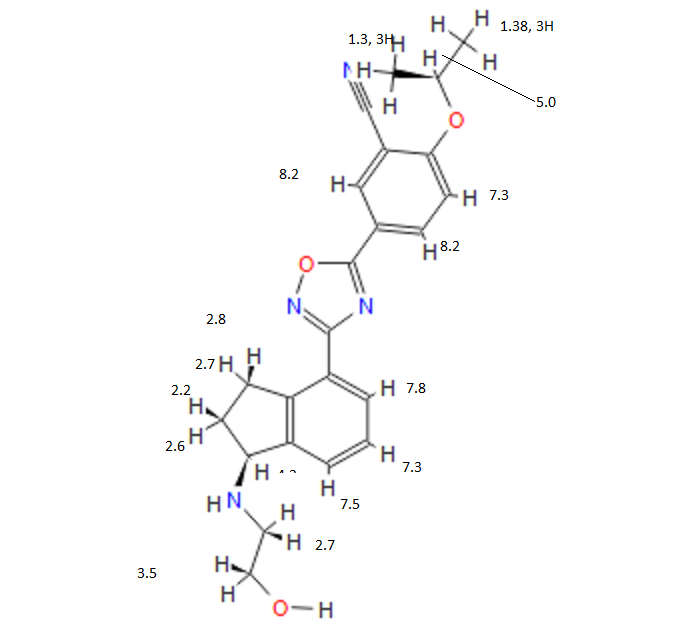
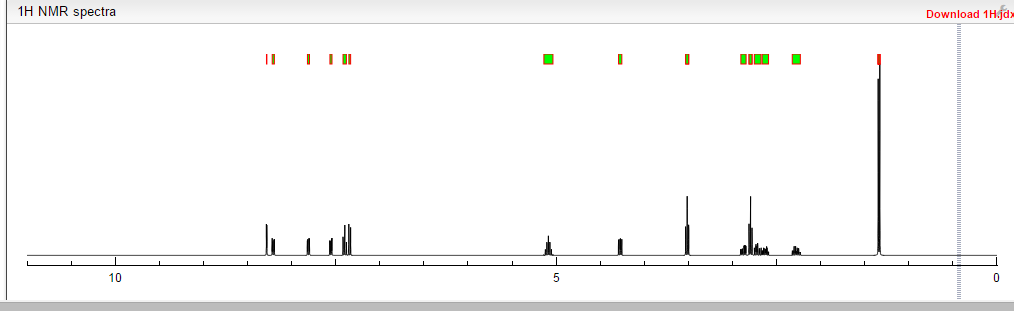
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_1h.png)
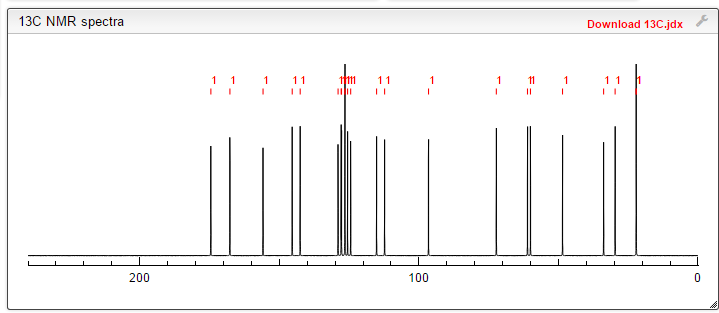
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_13c.png)
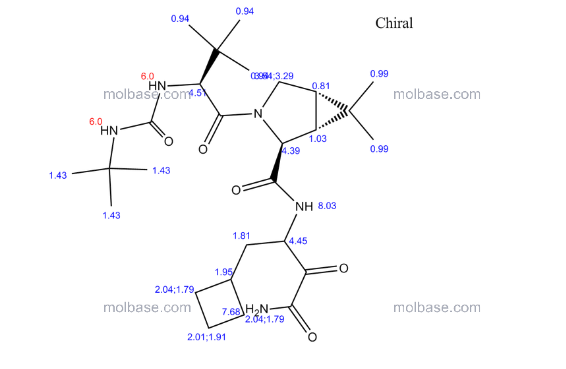
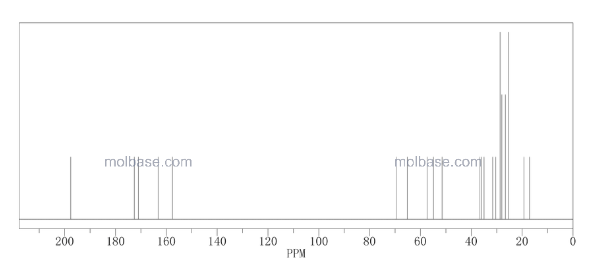

















































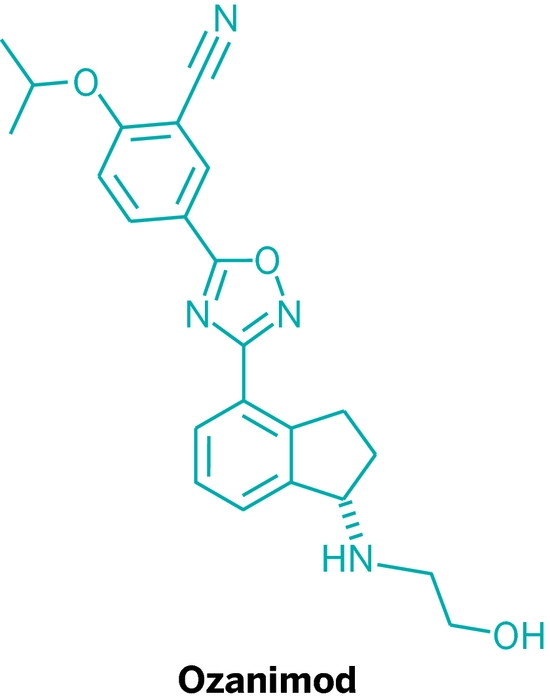

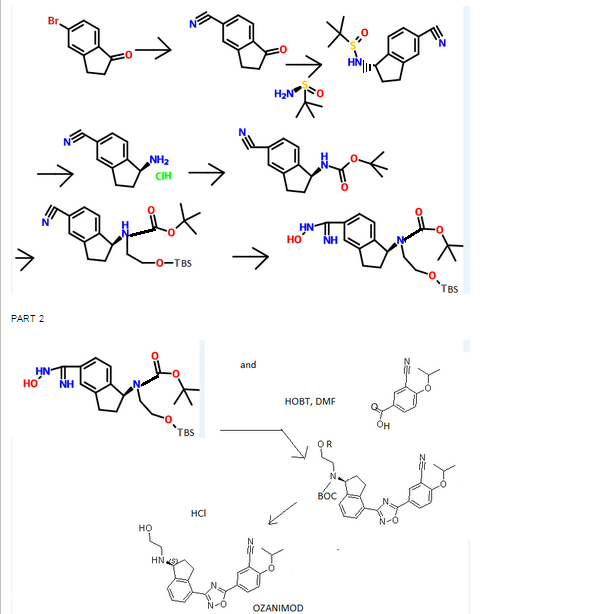





 CAS
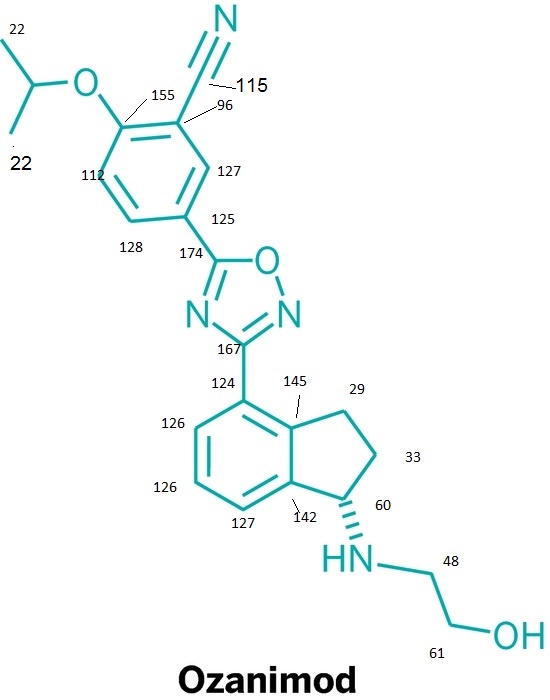
CAS 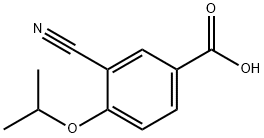 3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile
3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile (S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride
(S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride