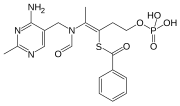
Benfotiamine
S-[(Z)-2-[(4-amino-2-methylpyrimidin-5-yl)methyl-formylamino]-5-phosphonooxypent-2-en-3-yl] benzenecarbothioate
Benphothiamine; Betivina; Biotamin; Neurostop; Nitanevril;22457-89-2
Benfotiamine (rINN, or S-benzoylthiamine O-monophosphate) is a synthetic S-acyl derivative of thiamine (vitamin B1).
It has been licensed for use in Germany since 1993 under the trade name Milgamma. (Combinations with pyridoxine or cyanocobalamin are also sold under this name.) It is prescribed there for treating sciatica and other painful nerve conditions.[1]
It is marketed as a medicine and/or dietary supplement, depending on the respective Regulatory Authority.[citation needed]



Uses
Benfotiamine is primarily marketed as an antioxidant dietary supplement. In a clinical study with six patients, benfotiamine lowered AGE by 40%.[2]
Benfotiamine may be useful for the treatment of diabetic retinopathy, neuropathy, and nephropathy however “Most of the effects attributed to benfotiamine are extrapolated from in vitro and animal studies. Unfortunately apparent evidences from human studies are scarce and especially endpoint studies are missing. Therefore additional clinical studies are mandatory to explore the therapeutic potential of benfotiamine in both diabetic and non-diabetic pathological conditions”.[3] It is thought that treatment with benfotiamine leads to increased intracellular thiamine diphosphate levels,[3] a cofactor of transketolase. This enzyme directs advanced glycation and lipoxidation end products (AGE’s, ALE’s) substrates to the pentose phosphate pathway, thus reducing tissue AGEs.[4][5][6][7][8]
Pharmacology
After absorption, benfotiamine can be dephosphorylated by cells bearing an ecto-alkaline phosphatase to the lipid-soluble S-benzoylthiamine.[9] Benfotiamine should not be confused with allithiamine, a naturally occurring thiamine disulfide derivative with a distinct pharmacological profile.[10]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=48F4CE7167F2EB243FBAF807987983D5.wapp1nB?docId=WO2014059702&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
The Benfotiamine, disclosed in US pat. no. 19623064000 US english names: S-benzoylthiamine O-monophosphate common name: Benfotiamine, chemical name: S − 2-[ [ (2-methyl-4-amino-5-pyrimidinyl) methyl ]-propionylamino ]-5-phosphonato-2-pentene-3-thiol benzoate, formula C 19 H 23 N 406 PS molecular weight 466.45 the following structural formula:

Chemical composition of the same species, in various physico-chemical conditions, crystallization into two or more different structure of the crystalline phenomenon, also referred to as polymorphs or homogeneous an image drug polymorph is a common phenomenon of drug discovery, drug quality is an important factor. Various polymorphs have different physical properties such as appearance, melting point, hardness, dissolution rate, chemical stability, mechanical stability, etc. differences, these differences in the physical properties of the sometimes affect the stability of the drug, bioavailability, even the drug availability. Thus, in drug development, it should be fully considered drug poly-type problems, the type of study and control in drug development of significant research content.
The benfotiamine, vitamin B 1 lipid-soluble derivatives, improved water-soluble vitamins B1 low bioavailability of disadvantages, increased blood and tissues. Thiamine concentration, thereby enhancing efficacy. The primary application to the following aspects (1) for thiamine deficiency disease prevention and treatment; (2) vitamin B 1 demand increases, from the food uptake is not sufficient make-up, fatigue, hyperthyroidism, gestation, lactation, vigorous manual labor, etc.); (3) for the treatment of non-l 酒性 lopinavir, grams of brain disease; (4) for the treatment of foot disease; (5) for the disease, the speculative and thiamine deficiency and metabolic disorders associated with treatment, such as: neuropathic pain; muscle pain, joint pain ; Peripheral-inflammatory, peripheral nerve
The paralysis; myocardial metabolism disorders, constipation, gastrointestinal motility dysfunction. The benfotiamine as vitamin B 1 supplemental agents have been in the united states, japan, europe, etc worldwide market. Recent studies have shown that, benfotiamine in diabetic peripheral neuropathy and retinopathy of significant therapeutic effect. In addition, our studies, benfotiamine may also be applied to the prevention and treatment of alzheimer’s disease, and aging.
Alzheimer’s disease (Altheimer’s disease, AD) is a cognitive, behavioral disorders is the primary clinical manifestations progressive neurodegenerative diseases, an age-related disorders, with age, their prevalence is a significant rise. 我国 the number of people in excess of 600 million AD patients, it is contemplated that in 2050 worldwide by the year AD patient may exceed 3000 million people as the medical scientific development, severe affect human health, mortality is a leading significant diseases such as cancer, stroke, cardiovascular disease, exhibit a decrease in mortality year by year, and AD mortality the rendering large increase in . In addition, alzheimer’s disease course long, the disabling rate is high, thus, alzheimer’s disease will be the 21 st century threaten both human diseases the most serious. It is estimated that worldwide by the year AD 2010 for medical costs up to 6040 of millions of dollars, the same global of the gross national product of 1%
China and the USA, the world there have been the following two classes of drugs approved for AD treatment: cholinesterase inhibitors and N-methyl D-aspartate (NMDA) receptor antagonist are both improved AD patient symptoms, slow disease progression does not prevent or reverse the progression of a disease. The benfotiamine by inhibiting the sugar synthase kinase -3 (Glycogen synthase kinase -3, GSK -3) activity, decrease in brain beta-amyloid protein (beta-amyloid, alpha beta) the deposition and tau protein phosphorylation, reduce alzheimer’s disease, pathological damage.
Presently available, benfotiamine primarily in the form of tablets and powders is administered in the form of, all formulations are not related to the benfotiamine feedstock form has not yet been the benfotiamine crystalline be systematically studied, the present US pat. no. first for benfotiamine of systematic study of various forms, illustrating different form benfotiamine characteristics and their feasibility. As a pharmaceutical agent

PATENT

http://www.google.com/patents/CN103772432A?cl=en
Example 1:
Was added to the reaction kettle 4000kg polyphosphoric acid, heated to 100 ~ 120 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range 8 hours, was added water quenching 3000kg off after the reaction, the temperature was raised to 80-90 ° C hydrolysis of 10 hours; cooled to room temperature, was added to the kettle 5000kg trioctylamine mixture of methyl tert-butyl ether = WPA / 1/1; aqueous phase 5000kg methanol to precipitate a solid, centrifuged to obtain a monoester 1200kg vitamin BI phosphoric acid crude; the 1200kg Vitamin `prime BI phosphate monoester crude in 6000kg water mixed beating, down to O ~ 5 ° C, dropping liquid in this temperature range adjusting the PH value of the base system to 12.0 ~ 14.0; PH after adjustment to ensure that the reactor temperature 10 ~ 25 ° C within 1200kg of benzoyl chloride was added dropwise, after the addition is complete heat the reaction to completion; filtered and the filtrate adjust PH from 3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1200kg, namely benfotiamine. Yield: 77.38%, Purity: 98.70% ο
Example 2:
Was added to the reaction kettle 5000kg polyphosphoric acid, heated to 80 ~ 100 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range 6 hours, was added water quenching 5000kg off after the reaction was heated to reflux for 5 hours hydrolysis; cooled to room temperature, the autoclave was added to the mixture was extracted twice 4000kg trioctylamine / methyl tert-butyl ether = 1/1; aqueous phase 6000kg ethanol precipitation The solid obtained by centrifugation vitamin BI phosphate monoester 1200kg crude; after 1200kg vitamin BI crude phosphate monoester product mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda adjust PH value system in this temperature range to 10.0 ~ 12.0; PH adjusting finished, to ensure the reactor temperature 10 ~ 25 ° C within 1200kg of benzoyl chloride was added dropwise, after the addition is complete heat the reaction to completion; filtered, the solid was filtered, the filtrate was adjusted to 3.5 ~ PH value 4.0 precipitated solid was isolated and dried to give a white solid 1250kg, namely benfotiamine. Yield: 80.61%, Purity: 98.50% ο
Example 3:
After the reactor was added 3000kg polyphosphoric acid, heated to 90 ~ 110 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after the insulation in this temperature range for 5 hours, 5000kg of water quenching off after the reaction, the temperature was raised to 90-100 ° C hydrolysis 5 hours; cooled to room temperature, was added to the kettle 5000kg trioctylamine methyl tert-butyl ether mixture was extracted twice = / 1/1; aqueous phase Join 7000kg acetone precipitate a solid, mono- 1230kg centrifuged to obtain crude vitamin BI phosphoric acid; vitamin BI after 1200kg crude phosphate monoester product mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda adjusted within this temperature range System PH value to 11.0 ~ 13.0; PH after adjustment to ensure that the temperature of the reactor was added dropwise within 10 ~ 25 ° C within 1200kg benzoyl chloride, and after the addition is complete heat to the completion of the reaction; filtered, the filtrate was adjusted to 3.5 PH value to 4.0 precipitated solid was isolated and dried to give a white solid 1240kg, namely benfotiamine. Yield: 79.96%, Purity: 98.50% ο
Example 4
Was added to the reaction kettle 4000kg polyphosphoric acid, heated to 100 ~ 120 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range for 4 hours, water quenching 8000kg off after the reaction, the temperature was raised to 90 – 110 ° C hydrolysis seven hours; cooled to room temperature, was added to the kettle 4000kg trioctylamine / methyl tert-butyl ether mixture was extracted phosphoric = 1/1; aqueous phase 6000kg methanol precipitated solid was centrifuged to give 1200kg vitamin BI phosphate monoester crude; the 1200kg vitamin BI phosphate monoester crude 6000kg water were mixed after beaten, cooled to O ~ 5 ° C, caustic soda was added dropwise at this temperature adjustment range of the system PH value to 9.0 ~ 11.0; PH adjustment finished, the reactor temperature to ensure solution of 10 ~ 25 ° C within 1200kg benzoyl chloride, and after the addition is complete heat to the completion of the reaction; filtered, the filtrate was adjusted to PH value
3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1260kg, namely benfotiamine. Yield: 81.24%, Purity: 98.70% ο
Example 5
Was added to the reaction kettle 5000kg polyphosphoric acid, heated to 110 ~ 130 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range for 3 hours, water quenching 10000kg off after the reaction, the temperature was raised to 110 – 120 ° C under reflux for 3 hours hydrolysis; cooled to room temperature, the mixture was extracted phosphoric acid was added to the kettle 3000kg trioctylamine / methyl tert-butyl ether = 1/1; aqueous phase `6000kg ethanol was added to precipitate a solid, obtained by centrifugation 1200kg vitamin BI phosphate monoester crude; after 1200kg vitamin BI phosphate monoester crude mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda in this temperature range adjusting the PH value of the system to the 8.0 ~ 10.0; PH adjusting finished, 1200kg of benzoyl chloride was added dropwise to ensure the kettle temperature within 10 ~ 25 ° C, after the addition is complete heat the reaction to completion; filtered, the filtrate was adjusted to PH value 3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1230kg, namely benfotiamine. Yield: 79.31%, purity: 98.60% ο
PATENT

http://www.google.com/patents/CN102911208A?cl=en
Example I: Phosphorus oxychloride 15. 33g (O. Imol) was added to the water 10. 8mL, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 26. 53g (O. lmol), warmed to 50 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 91.36%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, the 0_5 ° C under stirring, monitoring the reaction solution and pH changes, the pH value is stable, does not change when the reaction liquid PH, stirring was continued for I hour the reaction, the solution was adjusted to pH 3. 5-4. 0, suction filtration to give 33. 58g benfotiamine white solid. Yield 71.9%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, J = I. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 2: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 7. 2mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 21. 23g (O. OSmol), warmed to 60 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 92.37%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, suction filtration to give 27. 69g benfotiamine white solid. Yield 74.2%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3):.. 2.18 (s, 3H), 2 56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H ), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 3: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 3. 6mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 15. 92g (O. 06mol), warmed to 70 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 93.23%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, filtration, benfotiamine was a white solid 23. 71g. Yield 84.7%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 4: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 7. 2mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 10. 62g (O. 04mol), warmed to 80 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 95.26%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, filtration, benfotiamine was a white solid 15. 22g. Yield 85.2%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).

PATENT
http://www.google.com/patents/CN103724374A?cl=en
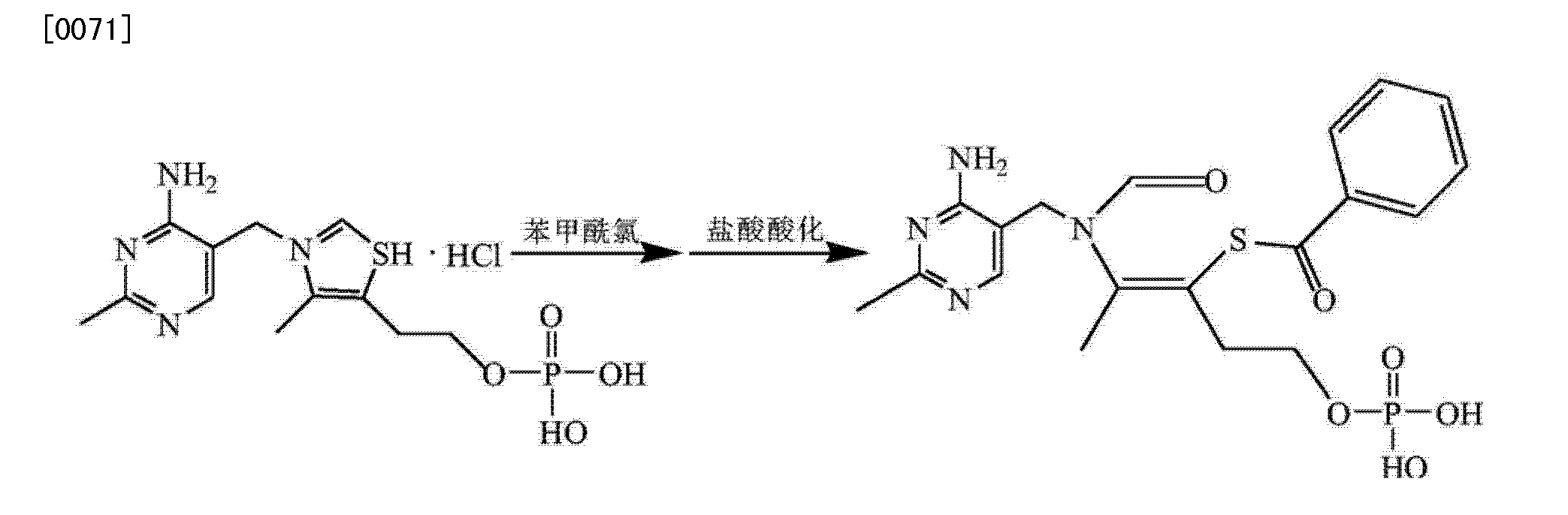
Synthesis I) thiamine monophosphate hydrochloride
In the reaction flask was added phosphate, thiamine hydrochloride, phosphorous pentoxide was added and stirred to dissolve, controlling the reaction temperature to complete the reaction thiamine hydrochloride, was added and stirring was continued after dropwise addition of concentrated hydrochloric acid hydrolysis of purified water was added dropwise acetone crystallization dropwise at raising grain, filtration, washed with acetone crystal, vacuum drying intermediates thiamine monophosphate hydrochloride;
2) Synthesis of crude benfotiamine
In the reaction flask thiamine monophosphate hydrochloride, dissolved in purified water, sodium hydroxide was added dropwise to adjust the pH to alkaline and steady, benzoyl chloride, sodium hydroxide was added dropwise while controlling alkaline pH, to control the temperature of the reaction pH remained stable, the end of the reaction, concentrated hydrochloric acid was added and extracted twice with ethyl acetate, the aqueous phase of sodium hydroxide was added dropwise until the pH is acidic, crystal seeding planting, filtration, purified water and acetone crystal, vacuum drying crude benfotiamine;
See also
References
- 1 “BBC news story: Back pain drug ‘may aid diabetics'”. BBC News. 18 February 2003.
- 2
- J Lin, A Alt, J Liersch, RG Bretzel, M Brownlee (May 2000). “Benfotiamine Inhibits Intracellular Formation of Advanced Glycation End Products in vivo” (PDF). Diabetes. 49 (Suppl1) (A143): 583.
- 3
- Balakumar P, Rohilla A, Krishan P, Solairaj P, Thangathirupathi A (2010). “The multifaceted therapeutic potential of benfotiamine”. Pharmacol Res 61 (6): 482–8. doi:10.1016/j.phrs.2010.02.008. PMID 20188835.
- 4
- Since AGEs are the actual agents productive of diabetic complications, in theory, if diabetic patients could block the action of AGEs completely by benfotiamine, strict blood sugar control, with its disruption of lifestyle and risks to health and life by severe hypoglycemic episodes, could be avoided, with revolutionary implications for the treatment of diabetes. Hammes, HP; Du, X; Edelstein, D; Taguchi, T; Matsumura, T; Ju, Q; Lin, J; Bierhaus, A; Nawroth, P; Hannak, D; Neumaier, M; Bergfeld, R; Giardino, I; Brownlee, M (2003). “Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy”. Nat Med 9 (3): 294–299. doi:10.1038/nm834.
- 5
- Stirban A, Negrean M, Stratmann B; et al. (2007). “Adiponectin decreases postprandially following a heat-processed meal in individuals with type 2 diabetes: an effect prevented by benfotiamine and cooking method”. Diabetes Care 30 (10): 2514–6. doi:10.2337/dc07-0302. PMID 17630265.
- 6
- Stracke H, Hammes HP, Werkmann D; et al. (2001). “Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats”. Exp. Clin. Endocrinol. Diabetes 109 (6): 330–6. doi:10.1055/s-2001-17399. PMID 11571671.
- 7
- Stirban A, Negrean M, Stratmann B; et al. (2006). “Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes”. Diabetes Care 29 (9): 2064–71. doi:10.2337/dc06-0531. PMID 16936154.
- 8
- Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ (2003). “Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine”. Diabetes 52 (8): 2110–20. doi:10.2337/diabetes.52.8.2110. PMID 12882930.
- 9
- Yamazaki, M (1968). “Studies on the absorption of S-benzoylthiamine O-monophosphate : (I) Metabolism in tissue homogenates”. Vitamins 38 (1): 12–20.
- 10
Volvert, M.L.; Seyen, S.; Piette, M.; Evrard, B.; Gangolf, M.; Plumier, J.C.; Bettendorff, L. (2008). “Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives”. BMC Pharmacology 8 (1): 10. doi:10.1186/1471-2210-8-10. PMC 2435522. PMID 18549472.
External links
| CN101654464A * |
Jul 28, 2009 |
Feb 24, 2010 |
湖北华中药业有限公司;湖北制药有限公司 |
Method for synthesizing vitamin B1 phosphatic monoester |
| CN102766163A * |
Jun 29, 2012 |
Nov 7, 2012 |
暨明医药科技(苏州)有限公司 |
Synthesis method of phosphate monoester of vitamin B1 |
| CN102911208A * |
Sep 25, 2012 |
Feb 6, 2013 |
同济大学 |
Method for synthesizing benfotiamine |
| CA682778A * |
|
Mar 24, 1964 |
Sankyo Kabushiki Kaisha |
S-benzoylthiamine o-monophosphate and a process for preparing the same |
| US3507854 * |
Apr 7, 1965 |
Apr 21, 1970 |
Sankyo Co |
Process for preparing thiamine derivatives |
| CN103772432A * |
Jan 3, 2014 |
May 7, 2014 |
湖北瑞锶科技有限公司 |
Production method of benfotiamine |
| CN103772432B * |
Jan 3, 2014 |
Jan 20, 2016 |
湖北瑞锶科技有限公司 |
一种苯磷硫胺的生产方法 |
| Patent |
Submitted |
Granted |
| Topical compositions comprising benfotiamine and pyridoxamine [US7666442] |
2006-03-02 |
2010-02-23 |
| METHODS OF USING BENFOTIAMINE AND PYRIDOXAMINE COMPOSITIONS [US2010151061] |
2010-06-17 |
|
| Topical delivery of trace metals for skin care [US7569558] |
2006-08-17 |
2009-08-04 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] |
2014-07-11 |
2014-10-30 |
| Protein Carrier-Linked Prodrugs [US2014323402] |
2012-08-10 |
2014-10-30 |
| ANTINEURITIC PHARMACEUTICAL COMBINATION AND COMPOSITIONS [US2014323428] |
2012-12-14 |
2014-10-30 |
| METHODS FOR IMPROVING MEDICAL THERAPIES [US2014335074] |
2012-12-13 |
2014-11-13 |
| LONG LASTING BREATH MINT [US2014335139] |
2014-05-13 |
2014-11-13 |
| High-Loading Water-Soluable Carrier-Linked Prodrugs [US2014296257] |
2012-08-10 |
2014-10-02 |
| PYRAZOLE-AMIDE COMPOUNDS AND PHARMACEUTICAL USE THEREOF [US2014296315] |
2014-03-14 |
2014-10-02 |
| Patent |
Submitted |
Granted |
| HYDRATE AND CRYSTAL OF FLUORENE COMPOUNDS [US2014296316] |
2014-03-14 |
2014-10-02 |
| Cysteine Peptide-Containing Health Drink [US2014302171] |
2012-10-11 |
2014-10-09 |
| Delaying the Progression of Diabetes [US2014303079] |
2012-05-08 |
2014-10-09 |
| FIBRONECTIN BASED SCAFFOLD DOMAIN PROTEINS THAT BIND TO MYOSTATIN [US2014309163] |
2014-05-12 |
2014-10-16 |
| METHODS AND COMPOSITIONS FOR CORRECTION OF ORGAN DYSFUNCTION [US2014274957] |
2014-03-13 |
2014-09-18 |
| COMPOUNDS FOR IMPROVED VIRAL TRANSDUCTION [US2014234278] |
2012-09-28 |
2014-08-21 |
| TOPICAL DERMAL DELIVERY COMPOSITIONS USING SELF ASSEMBLING NANOPARTICLES WITH CETYLATED COMPONENTS [US2014234428] |
2013-02-15 |
2014-08-21 |
| Polymeric Hyperbranched Carrier-Linked Prodrugs [US2014243254] |
2012-08-10 |
2014-08-28 |
| ENCAPSULATED OILS [US2014023688] |
2013-07-12 |
2014-01-23 |
| COMPOSITIONS, KITS AND METHODS FOR NUTRITION SUPPLEMENTATION [US2014023751] |
2013-09-27 |
2014-01-23 |
///////
O=P(O)(O)OCCC(/SC(=O)c1ccccc1)=C(/N(C=O)Cc2cnc(nc2N)C)C






















 CEO, YOUNG KIL CHANG
CEO, YOUNG KIL CHANG DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 .
. .
.