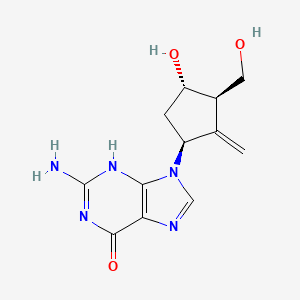
Forodesine
- Molecular FormulaC11H14N4O4
- Average mass266.253 Da

CAS No. : 284490-13-7
Molecular Formula: C11H15ClN4O4
Average Mass: 302.72 g/mol
Forodesine (INN; also known as Immucillin H; trade names Mundesine and Fodosine) is a transition-state analog inhibitor of purine nucleoside phosphorylase[1] studied for the treatment of patients with T-cell acute lymphoblastic leukemia (T-ALL) and for treatment of B-cell acute lymphocytic leukemia (B-ALL).
Forodesine was originally discovered by Vern Schramm‘s laboratory at the Albert Einstein College of Medicine in New York and Industrial Research Limited in New Zealand.
Forodesine is being developed by BioCryst Pharmaceuticals. As of 2008, it is currently in phase II clinical trials.[2].
In 2006, BioCryst entered into a licensing agreement with Mundipharma International Holdings Limited to develop and commercialize forodesine in markets across Europe, Asia, and Australasia for use in oncology.[3]
In April 2017, forodesine was approved in Japan for the treatment of relapsed/refractory peripheral T-cell lymphoma.[4]
ema
On 20 September 2010, orphan designation (EU/3/10/780) was granted by the European Commission to Mundipharma Research Limited, United Kingdom, for forodesine for the treatment of chronic lymphocytic leukaemia
| Active substance | Forodesine hydrochloride |
|---|---|
| Decision number | P/69/2010 |
| PIP number | EMEA-000785-PIP01-09 |
| Pharmaceutical form(s) | Hard capsule |
| Condition(s)/indication(s) | Cutaneous T-cell lymphoma (CTCL) |
| Route(s) of administration | Oral use |
| PIP applicant | Applicant: Mundipharma Research Ltd E-mail: [email protected] Country: United Kingdom Phone: +44 1223424900 Fax: +44 1223426054 |
| Decision type | W: decision granting a waiver in all age groups for the listed condition(s) |
On 20 September 2010, orphan designation (EU/3/10/780) was granted by the European Commission to Mundipharma Research Limited, United Kingdom, for forodesine for the treatment of chronic lymphocytic leukaemia.
What is chronic lymphocytic leukaemia?
Chronic lymphocytic leukaemia (CLL) is cancer of a type of white blood cell called B lymphocytes. In this disease, the lymphocytes multiply too quickly and live for too long, so that there are too many of them circulating in the blood. The cancerous lymphocytes look normal, but they are not fully developed and do not work properly. Over a period of time, the abnormal cells replace the normal white blood cells, red blood cells and platelets (components that help the blood to clot) in the bone marrow (the spongy tissue inside the large bones in the body). CLL is the most common type of leukaemia and mainly affects older people. It is rare in people under the age of 40 years. CLL is a long-term debilitating and life-threatening disease because some patients develop severe infections. What is the estimated number of patients affected by the condition? At the time of designation, CLL affected approximately 3 in 10,000 people in the European Union (EU)*. This is equivalent to a total of around 152,000 people, and is below the threshold for orphan designation, which is 5 people in 10,000. This is based on the information provided by the sponsor and the knowledge of the Committee for Orphan Medicinal Products (COMP).
What treatments are available? Treatment for CLL is complex and depends on a number of factors, including the extent of the disease, whether it has been treated before, and the patient’s age, symptoms and general state of health. Patients whose CLL is not causing any symptoms or is only getting worse very slowly may not need
Forodesine Hydrochloride was originally developed by BioCryst Pharmaceuticals and then licensed to Mundipharma and in particular is marketed in Japan under the trade name Mundesine®. Forodesine Hydrochloride is a transitional analogue inhibitor of purine nucleoside phosphorylase (PNP). Mundesine® is approved for the treatment of peripheral T-cell lymphoma (PTCL).
Mundesine® is a capsule that contains 100mg of free Forodesine per capsule. The recommended dose is 300mg orally, twice daily.
In 2004, the compound was eligible for orphan drug treatment for non-Hodgkin’s lymphoma (NHL), chronic myelogenous leukemia (CLL) and hairy cell leukemia, respectively. In 2007, the compound was eligible for the EU orphan drug for the treatment of acute lymphoblastic leukemia (ALL) and cutaneous T-cell lymphoma (CTCL). In 2010, the compound was eligible for EU orphan drug for treatment of CLL. In 2006, the compound obtained Japanese orphan drug eligibility for CTCL treatment.
Forodesine, or 7-[(2S,3S,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)-2-pyrrolidinyl]-l,5-dihydropyrrolo[2,3-e]pyrimidin-4-one, is an inhibitor of purine nucleoside phosphorylase. It is currently in development as a treatment for peripheral T-Cell Lymphoma .
W099/19338 describes a compound genus as a new class of inhibitors of nucleoside metabolism, including Forodesine. The compounds effect as inhibitors of purine nucleoside phosphorylase is taught as efficacious to suppress T-cell function and to treat infections caused by protozoan parasites.
WO00/61783 describes a number of processes for preparing molecules described in W099/19338. Reaction scheme 3 on page 23 of the published application describes a synthesis of Forodesine, characterised by the removal of two acid labile protecting groups in the final step to yield the hydrochloride salt.
Forodesine is a particularly difficult molecule to make on a commercial scale. The current process for manufacture requires a coupling reaction under cryogenic temperature conditions of -55C. Subsequent steps involve the use of a high pressure hydrogenation reaction. Such extreme reaction conditions provide for safety concerns, particularly when conducted on a bulk scale. Further the products of the reaction were extremely challenging to purify. The effect of all this is to require more sophisticated and expensive equipment at the manufacturing plant; all of which add up to an increased cost of goods for patients. Accordingly a new manufacturing process was sought.
Surprisingly a new route has been invented which is shorter, cheaper, less dangerous and provides an increased overall yield whilst still conforming to the required purity profile.
The current manufacturing process is described in Fig 1.
5C


, MeOH, reflux xchange
tallisation

Fig l
Within the diagram, the following acronyms are used, wherein NCS is N-Chlorosuccinimide, OTBDMS is t-butyldimethylsiloxy protecting group, MtBE is methyl t-butyl ether, (BOC)20 is di-t-butyldicarbonate and BOC is t-butyloxycarbonyl protecting group,
Particularly problematic in this process is the requirement to conduct the coupling of process step (iii) at exceptionally low temperature. Further challenges are provided by process step (v) the hydrogenation reaction to remove the benxylyoxymethyl (BOM) protecting group, before removing the other acid labile protecting groups.
Conducting hydrogenation reactions with their need for a high pressure environment requires specialist equipment. Such apparatus is expensive, adding to the cost of the materials produced. Despite the use of specialist equipment, safety concerns can never be eradicated. Whilst BOM can, in certain circumstances, be acid labile, treatment of analogues of the molecules described in Fig 1 with acid has always resulted in incomplete removal of the protecting group, leading to a large number of partially deprotected impurities. This makes purification exceptionally difficult as well as reducing the overall yield for the step.
A new improved process has been developed as described in Fig 2:
Toluene



Fig 2
The new route has a number of clear advantages. The coupling reaction (ix) is conducted at a warmer -15°C, rather than the challenging cryogenic conditions of -55°C required previously. It eradicates the hydrogenation step, avoiding the need for dangerous high pressure conditions. It also makes the overall process much quicker and cheaper; not only are the conditions challenging, but the reagents used in large quantities such as palladium are expensive and environmentally challenging.
The classical method to remove a BOM protecting group is by catalytic hydrogenation. It is however known to be unstable in acid conditions. For this reasons there have been previous attempts to remove BOM at the same time as the three acid labile protecting groups. This has always been unsuccessful as treatment with acid typically resulted in incomplete deprotection, leading to a mixture of products. This made for a tricky purification and a reduced yield. Surprisingly under the particular conditions described herein it has been possible to effect the transformation in greater yield and without a difficult purification. The final product is obtained in equal or greater purity than material obtained from the previous route.
PATENT
Scheme 13
HO OH 1 . HCI/Acetone, MeOH OCH,
2. PPh3, imidazole I
HO (EtO)2POCH2CN
OH O O
Ribose Λ 13a

References for preparation of compound 13a:
1. Mishra, Girija Prasad; Rao, Batchu Venkateswara; Tetrahedron: Asymmetry (2011), 22(7), 812-817.
2. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson,
James E; Organic Letters (2011), 13(7), 1594-1597.
3. WO 2010/085377 A2 (incorporated by reference).
4. Yadav, J. S.; Reddy, P. Narayana; Reddy, B. V. Subba; Synlett (2010), (3), 457- 461.
5. Song, Kai; Zheng, Guo-jun; Huaxue Shiji (2010), 32(2), 171-172.
6. Prabhakar, Peddikotla; Rajaram, Singanaboina; Reddy, Dorigondla Kumar;
Shekar, Vanam; Venkateswarlu, Yenamandra; Tetrahedron: Asymmetry (2010), 21(2), 216-221.
7. CN 101182342 A.
8. Baird, Lynton J.; Timmer, Mattie S. M.; Teesdale-Spittle, Paul H.; Harvey, Joanne
E; Journal of Organic Chemistry (2009), 74(6), 2271-2277.
9. Wang, Xiang-cheng; Wang, Gang; Qu, Gang-lian; Huaxue Shijie (2008), 49(4), 226-228.
10. Ivanova, N. A.; Valiullina, Z. R.; Shitikova, O. V.; Miftakhov, M. S; Russian
Journal of Organic Chemistry (2007), 43(5), 742-746.
11. Braga, Fernanda Gambogi; Coimbra, Elaine Soares; Matos, Magnum de Oliveira;
Lino Carmo, Arturene Maria; Cancio, Marisa Damato; da Silva, Adilson David; European Journal of Medicinal Chemistry (2007), 42(4), 530-537.
12. Wender, Paul A.; Bi, F. Christopher; Buschmann, Nicole; Gosselin, Francis; Kan, Cindy; Kee, Jung-Min; Ohmura, Hirofumi; Organic Letters (2006), 8(23), 5373- 5376.
13. Fei, Xiangshu; Wang, Ji-Quan; Miller, Kathy D.; Sledge, George W.; Hutchins, Gary D.; Zheng, Qi-Huang; Nuclear Medicine and Biology (2004), 31(8), 1033- 1041.
14. Abdel-Rahman, Adel A.-H.; Abdel-Megied, Ahmed E.-S.; Goda, Adel E.-S.; Zeid,
Ibrahim F.; El Ashry, El Sayed H; Nucleosides, Nucleotides & Nucleic Acids (2003), 22(11), 2027-2038.
15. Palmer, Andreas M.; Jager, Volker; European Journal of Organic Chemistry
(2001), (7), 1293-1308.
16. Paquette, Leo A.; Bailey, Simon; Journal of Organic Chemistry (1995), 60(24),
7849-56.
17. Classon, Bjoern; Liu, Zhengchun; Samuelsson, Bertil; Journal of Organic
Chemistry (1988), 53(26), 6126-30.
18. Kissman, Henry M.; Baker, B. R; Journal of the American Chemical Society
(1957), 79 5534-40.
References for cyclizations related to preparation of compounds of type 13d:
1. Davies, Stephen G.; Durbin, Matthew J.; Goddard, Euan C; Kelly, Peter M.;
Kurosawa, Wataru; Lee, James A.; Nicholson, Rebecca L.; Price, Paul D.;
Roberts, Paul M.; Russell, Angela J.; Scott, Philip M.; Smith, Andrew D; Organic & Biomolecular Chemistry (2009), 7(4), 761-776.
2. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul M.;
Russell, Angela J.; Savory, Edward D.; Smith, Andrew D.; Thomson, James E; Tetrahedron: Asymmetry (2009), 20(6-8), 758-772.
3. Davies, Stephen G.; Nicholson, Rebecca L.; Price, Paul D.; Roberts, Paul. M.;
Smith, Andrew D; Synlett (2004), (5), 901-903.
4. Brock, E. Anne; Davies, Stephen G.; Lee, James A.; Roberts, Paul M.; Thomson, James E; Organic Letters (2011), 13(7), 1594-1597.
5. Gary B. Evans, Richard H. Furneaux, Andrzej Lewandowicz, Vern L. Schramm, and Peter C. Tyler, Journal of Medicinal Chemistry (2003), 46, 3412-3423.
PATENT
WO 2016110527
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016110527


The invention also provides for the synthesis of a compound of formula (II)

By reacting a compound of Formula (VII)

With di-t-butyldicarbonate.
Preferably the reaction is conducted at -10 to -20°C, in methyl t-butyl ether & heptane
The invention also provides for the synthesis of a compound of formula (VII)

By reacting a compound of Formula (IV)

With a suitable base to form

Before reacting with a compound of Formula (III)

Example 1
Stage 1 Manufacture of (III)
Compound of formula (III) (approx. 130g) in toluene solution is added to a suspension of N-Chlorosuccinimide in toluene at 20°C over a period of 90min. The reaction mixture is stirred at 20°C for 1 hour then chilled to 0°C and stirred for a further hour. The precipitated succinimide by-product is removed by filtration and the filtered solution charged directly to a 45% potassium hydroxide solution (aq) containing
tetrabutylammonium bromide. The reaction mixture is stirred at 0°C and completion of reaction is confirmed by GC analysis. Water is then added to the two-phase mixture to dissolve inorganic precipitates and the toluene product solution is washed with a 28% ammonium hydroxide/acetic acid buffer mixture with sodium chloride added. After phase separation the organic phase solution is stabilised with triethylamine. Magnesium sulfate is added to dry the solution. After filtration, the yield of (III) is determined by R.O.E. and GC purity.
Stage 2 Manufacture of (II)
Stage 2a Lithiation
A suspension of compound of formula (IV) (approx. 200g) in MtBE is chilled to -15°C and treated with /7-Hexyl lithium (2.5M in hexanes) added over 2h, maintaining the reaction mixture at -15°C. The mixture is then stirred for 3h at -15°C.
Stage 2b Coupling with (IV)
After lithiation is complete, a compound formula (III) in toluene solution is added to the reaction mixture maintaining the contents at -15°C. The reaction mixture is then stirred at this temperature for 1.5h.
Stage 2c Boc anhydride quench
A solution of di-t-butyldicarbonate in MtBE is added to the above reaction mixture at -15°C. The solution is stirred for a further 30min.
Workup and Purification
The reaction mixture is quenched by addition of RO water, then filtered. The aqueous layer is separated and run to waste. The organic layer is again washed with water. The organic layer is concentrated to a low volume and solvent replaced by heptane. The mix is stirred for 16h and filtered again.
The solution is passed through a silica gel column and eluted with heptane. The resulting solution is treated with charcoal – stirred for 3h, then filtered. The product (II) is progressed as a solution in heptane to the next stage.
Stage 3 Manufacture of Crude Forodesine (la)
Stage 3 Deprotection with cone. HCI
Concentrated hydrochloric acid is added to (II) in heptane and the mixture stirred. The acid phase is separated off and stirred for 16h at ambient temperature. The solution is then heated to 40°C for 6h. The water is then distilled off under reduced pressure to a minimum volume.
Ethanol is then added to precipitate the crude Forodesine (la) which is isolated by filtration after cooling 0-5°C. It is washed with ethanol and dried in a vacuum oven at 75°C to a constant weight.
Stage 4a Decolourization of crude Forodesine (la) using Ion-Exchange Column
Crude Forodesine (la) is dissolved in water and loaded onto a freshly prepared ion-exchange column containing Dowex 50WX4 resin in the Na+ form activated with 30% sodium hydroxide solution. The ion-exchange column is eluted with 4 x lOOmL water followed by 4 x lOOmL 2M HCI. The HCI fractions are collected separately as they contain the desired product. The 2M HCI fractions are combined and concentrated under vacuum with minimum RO water added to dissolve the residue. 1,4-Dioxane is added to the aqueous solution to precipitate the product. The mixture is stirred at 20°C for 1.5h. The product is filtered, washed with 1,4-dioxane and dried in a vacuum oven at 35°C to a constant weight to give decolourised BCX1777.
Stage 4b Recrystallization of Forodesine
Decolourised Forodesine is added to in 0.6M dilute hydrochloric acid and heated to 45°C to dissolve. The resulting solution is hot filtered and washed through with some RO Water. The solution is cooled to 20°C and ethanol added over at least lh. The mixture is then seeded with Forodesine HCI. The resulting slurry is stirred for 8h at 20°C, then cooled to 2°C for a further 1.5h. The product is isolated by filtration, washed twice with cold ethanol then dried in a vacuum oven at 75°C to a constant weight to give a white crystalline Forodesine HCI (approx. 50g).
While the invention has been described in detail and with reference to specific embodiments thereof, it will be apparent to one of skill in the art that various changes and modifications can be made therein without departing from the spirit and scope thereof. Moreover, all embodiments described herein are considered to be broadly applicable and combinable with any and all other consistent embodiments, as appropriate.
PAPER
Journal of Medicinal Chemistry (2009), 52(4), 1126-1143.
Third-Generation Immucillins: Syntheses and Bioactivities of Acyclic Immucillin Inhibitors of Human Purine Nucleoside Phosphorylase
* To whom correspondence should be addressed. Phone: +64-4-9313040. Fax: +64-4-9313055. E-mail: [email protected]., †
Carbohydrate Chemistry Team, Industrial Research Limited.
, ‡
Department of Biochemistry, Albert Einstein College of Medicine of Yeshiva University.

References
- Jump up^ Kicska GA, Long L, Hörig H, et al. (April 2001). “Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes”. Proc. Natl. Acad. Sci. U.S.A. 98 (8): 4593–8. Bibcode:2001PNAS…98.4593K. doi:10.1073/pnas.071050798. PMC 31879
 . PMID 11287638.
. PMID 11287638. - Jump up^ “Complete list of clinical trials for forodesine (BCX-1777) (ClinicalTrials.gov)”. Retrieved 2008-07-22.
- Jump up^ “Biocryst Initiates Pivotal Fodosine Phase IIb Clinical Trial In Patients With Relapsed/Refractory T-Lymphoblastic Leukemia/Lymphoma”(Press release). January 16, 2007.
- Jump up^ “BioCryst Announces Mundipharma Receives Approval for Mundesine® in Japan” (Press release). April 3, 2017.
External links
- “From cell biology to therapy: forodesine”. Hematology Meeting Reports. 2 (5): 106–111. 2008.
- Gore, L; Stelljes, M; Quinones, R (2007). “Forodesine treatment and post-transplant graft-versus-host disease in two patients with acute leukemia: Facilitation of graft-versus-leukemia effect?”. Seminars in Oncology. 34 (6 Suppl 5): S35–9. doi:10.1053/j.seminoncol.2007.11.005. PMID 18086346.
- 18 December 2006 Fodosine orphan designation by the European Commission for acute lymphoblastic leukaemia.
- BioCryst Pharmaceuticals, Inc. have entered into an exclusive license agreement with Mundipharma for develop and commercialize BioCryst’s lead compound, Forodesine.
- Birmingham, Alabama – February 2, 2006 Mundipharma will obtain rights in markets across Europe, Asia and Australasia to Forodesine™ in the field of oncology in exchange for a $10 million up-front payment. Furthermore, Mundipharma will commit up to an additional $15 million to assist in the evaluation of Forodesine’s™ therapeutic safety and efficacy profile. BioCryst may also receive future event payments totalling $155 million in addition to royalties on product sales of Forodesine™ by Mundipharma.
- News BioCryst provides Fodosine update March 27, 2007. “Voluntarily Placed on Hold by BioCryst (…) we don’t think the final response rate will be as high as 18%”.
- The European Commission granted a marketing authorisation valid throughout the European Union for Atriance on 22 August 2007 for acute lymphoblastic leukaemia. What benefit has Atriance shown during the studies? Atriance was shown to be effective in a proportion of the patients in both studies. In the first study, among the 39 children and young adults who se cancer had not responded to two or more previous treatments, five (13%) had a complete response to treatment after a month, with no evidence of disease and normal blood counts. In the second study, among the 28 adults and adolescents with cancer that had not responded to two or more previous tre atments, five (18%) had a complete response to treatment. In both studies, more patients had a partial response to Atriance treatment, with blood counts returning towards normal levels.
- Lino Berton collects all the information on Forodesine in www.linoberton.com site, putting them in a row. In 2014 he published the book Qualcosa che non muore where he tells his incredible experience in the closed trial early in 2007.
- Il Giornale.it (in Italian). “Come si boicotta un farmaco che funziona”. Dated 08-01-2016.
 |
|
| Clinical data | |
|---|---|
| Trade names | Mundesine and Fodosine |
| Routes of administration |
oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H14N4O4 |
| Molar mass | 266.26 g·mol−1 |
| 3D model (JSmol) | |
/////////Forodesine Hydrochloride, Forodesine, BCX 1777, I










































![2D [1H,1H]-TOCSY, 7.4 spectrum for 4-Aminophenol](https://i0.wp.com/www.bmrb.wisc.edu/ftp/pub/bmrb/metabolomics/entry_directories/bmse000462/nmr/set01/spectra/HH_TOCSY.png?w=695)



![2D [1H,13C]-HSQC, 7.4 spectrum for 4-Aminophenol](https://i0.wp.com/www.bmrb.wisc.edu/ftp/pub/bmrb/metabolomics/entry_directories/bmse000462/nmr/set01/spectra/1H_13C_HSQC.png?w=695)
![2D [1H,13C]-HMBC, 7.4 spectrum for 4-Aminophenol](https://i0.wp.com/www.bmrb.wisc.edu/ftp/pub/bmrb/metabolomics/entry_directories/bmse000462/nmr/set01/spectra/1H_13C_HMBC.png?w=695)