












Dr Anthony’s New Drug Approvals hits 13 lakh views in 212 countries

An Indian helping millions

MAKING INDIANS FEEL PROUD
LINK
////////blog, Dr Anthony , New Drug Approvals, 13 lakh views, 212 countries, India
Dr Anthony’s New Drug Approvals hits 13 lakh views in 212 countries
An Indian helping millions
MAKING INDIANS FEEL PROUD
LINK
////////blog, Dr Anthony , New Drug Approvals, 13 lakh views, 212 countries, India

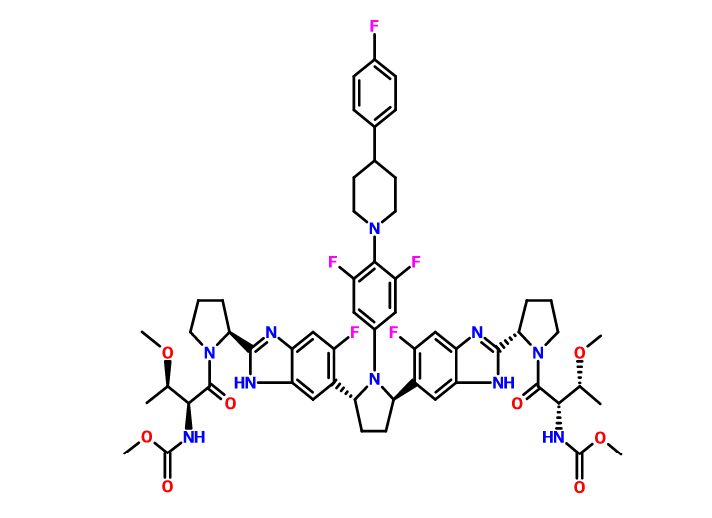
ABT-530, Pibrentasvir, A 1325912.0
Dimethyl N,N’-([(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}pyrrolidine-2,5-diyl]bis{(6-fluoro-1H-benzimidazole-5,2-diyl)[(2S)-pyrrolidine-2,1-diyl][(2S,3R)-3-methoxy-1-oxobutane-1,2-diyl]})biscarbamate
Methyl {(2S,3R)-1-[(2S)-2-{5-[(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}-5-(6-fluoro-2-{(2S)-1-[N-(methoxycarbonyl)-O-methyl-L-threonyl]pyrrolidin-2-yl}-1H-benzimidazol-5-yl)pyrrolidin-2-yl]-6-fluoro-1H-benzimidazol-2-yl}pyrrolidin-1-yl]-3-methoxy-1-oxobutan-2-yl}carbamate
Phase III
Abbott Laboratories INNOVATOR
A protease inhibitor potentially for the treatment of HCV infection.
Hepatitis C virus NS 5 protein inhibitors
![]()
CAS No. 1353900-92-1
| MF | C57H65F5N10O8 |
|---|
MW 1113.1925 MW
Pibrentasvir
SYNTHESIS
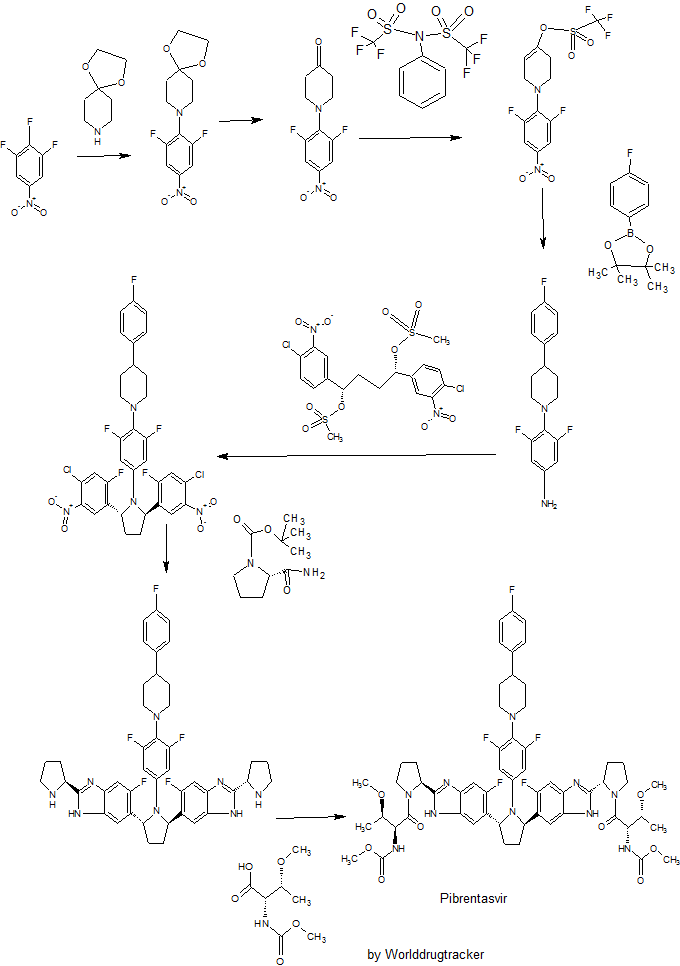
PATENT
WO 2012051361
http://www.google.com/patents/WO2012051361A1?cl=en

Example 3.52 methyl {(2S,3R)-l-[(2S)-2-{5-[(2R,5R)-l-{3,5-difluoro-4-[4-(4- fluorophenyl)piperidin-l-yl]phenyl}-5-(6-fluoro-2-{(2.S)-l-[A^-(methoxycarbonyl)-0-methyl-L- threonyl]pyiTolidin-2-yl}-l f-benzimidazol-5-yl)pyiTolidin-2-yl]-6-fluoro-l f-benzimidaz yl}pyrrolidin-l-yl]-3-methoxy-l-oxobutan-2-yl}carbamatelH NMR (400 MHz, DMSO) δ 12.36 – 12.06 (m, 2H), 7.41 (dd, J = 11.2, 6.3, 1H), 7.34 (dd, J = 10.4, 4.8, 1H), 7.30 – 7.20 (m, 3H), 7.17 – 6.98 (m, 5H), 5.98 – 5.82 (m, 2H), 5.65 – 5.47 (m, 2H), 5.17 – 5.06 (m, 2H), 4.25 (dd, J = 15.6, 8.1, 2H), 3.88 – 3.74 (m, 3H), 3.53 (d, J = 1.3, 6H), 3.49 – 3.38 (m, 2H), 3.31 (d, 1H), 3.25 (d, J = 3.7, 1H), 3.13 (d, J = 1.3, 3H), 3.03 (d, J = 2.3, 3H), 3.00 – 2.84 (m, 3H), 2.60 – 2.53 (m, J = 2.5, 2H), 2.26 – 1.55 (m, 14H), 1.28 – 1.13 (m, 1H), 1.10 – 0.88 (m, 6H). MS (ESI; M+H) m/z = 1113.4.

Intermediate 5
( 15,45)- 1 ,4-bis(4-chloro-3 -nitrophenyl)butane- 1 ,4-diyl dimethanesulfonate Intermediate 5A
2-bromo- 1 -(4-chloro-3 -nitrophenyl)ethanone
Method A:
To a flask equipped with a magnetic stir bar and under an atmosphere of N2 was added 4′- chloro-3 ‘-nitroacetophenone (10.0 g, 50.1 mmol) and THF (100 mL). To this stirring mixture was added portion-wise phenyltrimethylammonium tribromide (19.78 g, 52.6 mmol) over a 15 minutes time period. The resultant mixture was then stirred with monitoring every hour via LCMS. After 3 hours, the mixture was then filtered and resulting solids washed with EtOAc. The organic solution was then concentrated, and 10% aq. NaHCC^ were added, and the mixture was washed with EtOAc (2×300 mL). The combined organic layers were then washed with brine, dried (MgSO^), filtered and concentrated. The residue material was then subjected to purification via crystallization. The residue was dissolved in EtOAc (100 mL) and hexanes were slowly added until the mixture was cloudy. After standing for a few hours, 2-bromo- l-(4-chloro-3-nitrophenyl)ethanone (9.81 g, 70%) was collected as an off white colored solid product. !H NMR (500 MHz, DMSO-cfe) δ ppm 5.00 (s, 2 H) 7.98 (d, J=8.54 Hz, 1 H) 8.24 (dd, J=8.54, 2.14 Hz, 1 H) 8.61 (d, J=1.98 Hz, 1 H).
Method B:
In a 500 mL round-bottomed flask was added l -(4-chloro-3-nitrophenyl)ethanone (1 1.98 g, 60 mmol) in benzene (75 mL) to give a white suspension. Bromine (9.59 g, 60.0 mmol) was added dropwise over 5 minutes to give a deep red solution. The mixture was stirred for 1 hour to give a yellow solution that was concentrated in vacuo to a yellow solid. Recrystallized from 9: 1 hexane/ethyl acetate gave 2-bromo-l -(4-chloro-3-nitrophenyl)ethanone as yellow needles.

Intermediate 21
(1 S,4S)-1 -(4-chloro-2-fluoro-5 -nitrophenyl)-4-(4-chloro-3 -nitrophenyl)butane- 1 ,4-diyl
dimethanesulfonate
Intermediate 21 can be made from Intermediate 20B and l-(4-chloro-3-nitrophenyl)ethanone (commercially available from Aldrich) following the general methods to prepare Intermediate 20E.

l-(2,6-difluoro-4-nitrophenyl)piperidin-4-one
The crude 8-(2,6-difluoro-4-nitrophenyl)-l,4-dioxa-8-azaspiro[4.5]decane from the preceding procedure was dissolved in 4:1 acetone:water (100 mL). Concentrated HC1 (5 mL) was added, and the resulting mixture was stirred at 50 °C for 8 hours and then cooled to room temperature. The mixture was concentrated in vacuo to approximately 20 mL, which was carefully added to concentrated aq. NaHCC^ (100 mL) and extracted with EtOAc (2 x 100 mL). The combined organic extracts were dried over Na2S04, filtered and concentrated in vacuo. The crude product was triturated with Et20 and hexanes to give a bright-yellow solid that was collected and dried to provide the title compound (7.13 g, 81%).
PATENT
The present invention features crystalline polymorphs of methyl {(2S,3R)-1- [(2S)-2-{5-[(2R,5R)-l-{3,5-difluoro-4 4-(4-fluorophenyl)piperidin-l-yl]phenyl}-5-(6-fluoro-2-{(2S)- 1 -[N-(methoxycarbonyl)-0-methyl-L-threonyl]pyrrolidin-2-yl} – 1 H-benzimidazol-5-yl)pyrrolidin- -yl] -6-fluoro- 1 H-benzimidazol-2-yl} pyrrolidin- 1 -yl] -3 -methoxy- 1 -oxobutan-2-
yl} carbamate 
, herein “Compound I”). Compound I is a potent HCV NS5A inhibitor and is described in U.S. Patent Application Publication No. 2012/0004196, which is incorporated herein by reference in its entirety.
//////////1353900-92-1, PHASE 3, ABT-530, Pibrentasvir, ABT 530, A 1325912.0
C[C@H]([C@@H](C(=O)N1CCC[C@H]1c2[nH]c3cc(c(cc3n2)[C@H]4CC[C@@H](N4c5cc(c(c(c5)F)N6CCC(CC6)c7ccc(cc7)F)F)c8cc9c(cc8F)[nH]c(n9)[C@@H]1CCCN1C(=O)[C@H]([C@@H](C)OC)NC(=O)OC)F)NC(=O)OC)OC
C[C@H]([C@@H](C(=O)
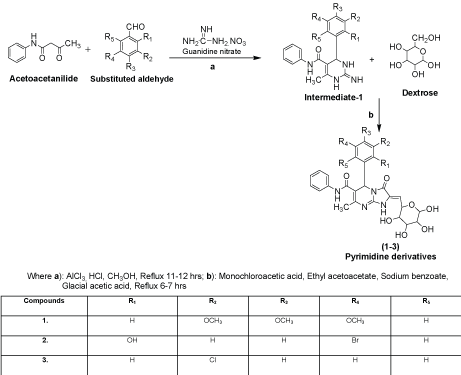
Procedure for the synthesized derivatives (1-3)
Step 1: The synthesis of Intermediate-1 (2-imino-6-methyl-4- (substituted aldehyde)-N-phenyl-1,2,3,4-tetrahydropyrimidine-5- carboxamide): A mixture of substituted aldehyde (0.01 mol), guanidine nitrate (0.015 mol) and acetoacetanilide (0.01 mol) were placed in round bottom flask in methanol (50 ml) and added aluminum chloride (0.003 mol) with 2-3 drops of conc. hydrochloric acid as catalyst amount then the reaction mixture was refluxed for 11-12 hrs after that the reaction mixture was cooled to room temperature and poured into crushed ice water with vigorous stirring, filtered and recrystallized with methanol [10].
Step 2: The synthesis of 5-(substituted aldehyde)-7-methyl-3-oxo-N-phenyl-2-((3,4,5,6-tetrahydroxy-tetrahydro-2H-pyran- 2-yl)methylene)-1,2,3,5-tetrahydroimidazo[1,2-a] pyrimidine-6- carboxamide derivatives: A mixture of intermediate-1 (0.01 mol), sodium benzoate (2 g), dextrose (0.01 mol), glacial acetic acid (20 ml), ethyl acetoacetate (15 ml) and monochloroacetic acid (0.015 mol) were taken in RBF and refluxed with controlled temperature at 140-142°C for 6-7 hrs. The reaction mixture was cooled at room temperature and poured into ice cold water to yielded solid precipitate or titled compounds (1-3), filtered and recrystallized with methanol.
Compound 1 (7-Methyl-3-oxo-N-phenyl-2-((3,4,5,6- tetrahydroxy-tetrahydro-2H-pyran-2-yl)methylene)-5-(3,4,5- trimethoxyphenyl)-1,2,3,5-tetrahydroimidazo[1,2-a]pyrimidine-6- carboxamide): IR (KBr pellets, cm-1): 3062 (C-H str., phenyl nucleus), 1596 (C=C str., phenyl nucleus), 694 (C-C str., phenyl nucleus), 1630 (C=O str.,), 3321 (N-H str., 2˚amide), 1630 (N=CH str., pyrimidine), 1244 (C-N str., pyrimidine), 2779 (C-H str., cyclic ether), 1126 (C-O-C str., aryl ether), 3321 (O-H str., polyhydroxy on dextrose), 1244 (C-O-C str., OCH3); 1H NMR (DMSO-d6, δppm): 7.45-7.49 (m, 7H, Ar-H), 2.10 (s, 1H, NH, amide), 8.25 (s, 1H, NH, amide), 3.86-4.22 (m, 5H, CH, tetrahydropyran), 2.10 (m, 4H, OH alcohol), 3.86 (m, 9H, OCH3).
| Jyoti Rani, Monika Saini, Sanjiv Kumar and Prabhakar Kumar Verma* | |
| Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak-124 001, Haryana, India | |
| *Corresponding Author : | Prabhakar Kumar Verma Department of Pharmaceutical Sciences Maharshi Dayanand University Rohtak-124 001, Harayana, India Tel: 9992581437 E-mail: vermapk422@rediffmail.com |
| Received March 07, 2016; Accepted March 29, 2016; Published March 31, 2016 | |
| Citation: Rani J, Saini M, Kumar S, Verma PK (2016) Synthesis, Biological Evaluation and Validation Studies of Novel 5-(Substituted Aldehyde)-2-imino-7-methyl-3-oxo-N-phenyl-1,2,3,5-tetrahydroimidazo[1,2-a]pyrimidine-6-carboxamide Scaffolds. Med chem (Los Angeles) 6:218-223. doi:10.4172/2161-0444.1000349 | |
| Copyright: © 2016 Rani J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. | |
link
///////////// 5-(Substituted Aldehyde)-2-imino-7-methyl-3-oxo-N-phenyl-1,2,3,5-tetrahydroimidazo[1,2-a]pyrimidine-6-carboxamide, Scaffolds
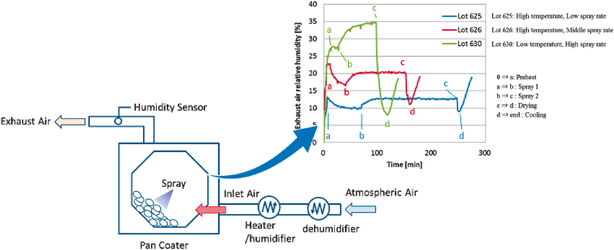
In the pharmaceutical tablet film coating process, we clarified that a difference in exhaust air relative humidity can be used to detect differences in process parameters values, the relative humidity of exhaust air was different under different atmospheric air humidity conditions even though all setting values of the manufacturing process parameters were the same, and the water content of tablets was correlated with the exhaust air relative humidity. Based on this experimental data, the exhaust air relative humidity index (EHI), which is an empirical equation that includes as functional parameters the pan coater type, heated air flow rate, spray rate of coating suspension, saturated water vapor pressure at heated air temperature, and partial water vapor pressure at atmospheric air pressure, was developed. The predictive values of exhaust relative humidity using EHI were in good correlation with the experimental data (correlation coefficient of 0.966) in all datasets. EHI was verified using the date of seven different drug products of different manufacturing scales. The EHI model will support formulation researchers by enabling them to set film coating process parameters when the batch size or pan coater type changes, and without the time and expense of further extensive testing.
EHI is defined as the following equation:
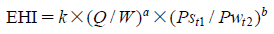 |
In general, pharmaceutical film coatings are applied in order to protect core tablets from light or for masking the taste of the active pharmaceutical ingredients. Therefore, the surface state of the coating layer is important to maintain the expected performance. During the coating process, however, the coating layer surface state is affected by the water content of the tablets. In a conventional approach, the water content of drug products is maintained at the validated level by monitoring the product’s temperature and/or the exhaust air temperature during the coating process. In a scale up study, the batch scale and manufacturing equipment are changed according to the progress of the process development stage. At each stage, the water content of drug products is constantly monitored and well-controlled to secure the consistency of the drug product’s quality. In this approach, numerous experiments are necessary to optimize the process parameters in each batch scale. As a result, the costs of materials, human resources, and time for development will become considerable.
In this study, the relationship between film coating process parameters and EARH was clarified. In addition, it was confirmed that the EARH affected the water content of tablets. These results indicated that the water content of tablets can be regulated by controlling the EARH. From these results, we proposed the EHI for quantification of the pharmaceutical film coating process. The fitting parameters in the EHI equation were set using the experimental data of 10 drug products and 7 kinds of pan coaters. These fitting parameters of EHI were validated by evaluating the correlation coefficient determined by comparing the calculated values of EARH and the measured experimental values of EARH from various drug products, pan coater scales and coating parameters. The main advantage of the EHI method is that commercial scale coating conditions can be predicted using only one film coating experimental result from a lab-scale pan coater.
/////////pan coater, exhaust air relative humidity index (EHI), scale up, drying ability, atmospheric air, tablet water content
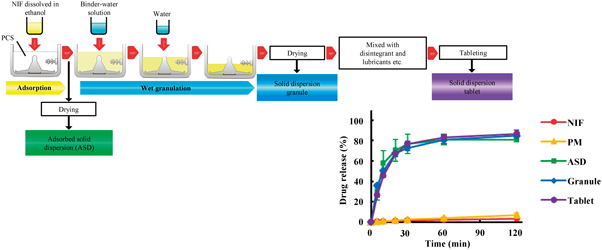
The aim of this study was to prepare and evaluate solid dispersion tablets containing a poorly water-soluble drug using porous calcium silicate (PCS) by a wet granulation method. Nifedipine (NIF) was used as the model poorly water-soluble drug. Solid dispersion tablets were prepared with the wet granulation method using ethanol and water by a high-speed mixer granulator. The binder and disintegrant were selected from 7 and 4 candidates, respectively. The dissolution test was conducted using the JP 16 paddle method. The oral absorption of NIF was studied in fasted rats. Xylitol and crospovidone were selected as the binder and disintegrant, respectively. The dissolution rates of NIF from solid dispersion formulations were markedly enhanced compared with NIF powder and physical mixtures. Powder X-ray diffraction (PXRD) confirmed the reduced crystallinity of NIF in the solid dispersion formulations. Fourier transform infrared (FT-IR) showed the physical interaction between NIF and PCS in the solid dispersion formulations. NIF is present in an amorphous state in granules prepared by the wet granulation method using water. The area under the plasma concentration–time curve (AUC) and peak concentration (Cmax) values of NIF after dosing rats with the solid dispersion granules were significantly greater than those after dosing with NIF powder. The solid dispersion formulations of NIF prepared with PCS using the wet granulation method exhibited accelerated dissolution rates and superior oral bioavailability. This method is very simple, and may be applicable to the development of other poorly water-soluble drugs.
Solid dispersion formulations of NIF with PCS using the wet granulation method were prepared and evaluated. These formulations exhibited much higher dissolution rates than NIF powder, comparable to ASD. Furthermore, these formulations provided superior bioavailability in rats compared with NIF powder. NIF was present in the amorphous state in the granules after preparation by a wet granulation method using water. The wet granulation method proposed here is very simple, and may be applicable to other poorly water-soluble drugs.
Solid dispersion formulations of NIF with PCS using the wet granulation method were prepared and evaluated. These formulations exhibited much higher dissolution rates than NIF powder, comparable to ASD. Furthermore, these formulations provided superior bioavailability in rats compared with NIF powder. NIF was present in the amorphous state in the granules after preparation by a wet granulation method using water. The wet granulation method proposed here is very simple, and may be applicable to other poorly water-soluble drugs.

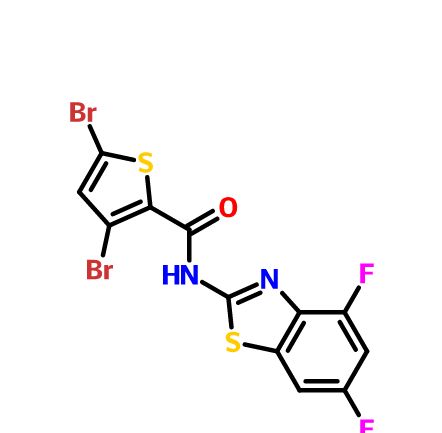
New and novel anti-norovirus agents
There is an urgent need for structurally novel anti-norovirus agents. In this study, we describe the synthesis, anti-norovirus activity, and structure–activity relationship (SAR) of a series of heterocyclic carboxamide derivatives. Heterocyclic carboxamide 1 (50% effective concentration (EC50)=37 µM) was identified by our screening campaign using the cytopathic effect reduction assay. Initial SAR studies suggested the importance of halogen substituents on the heterocyclic scaffold and identified 3,5-di-boromo-thiophene derivative 2j (EC50=24 µM) and 4,6-di-fluoro-benzothiazole derivative 3j (EC50=5.6 µM) as more potent inhibitors than 1. Moreover, their hybrid compound, 3,5-di-bromo-thiophen-4,6-di-fluoro-benzothiazole 4b, showed the most potent anti-norovirus activity with a EC50 value of 0.53 µM (70-fold more potent than 1). Further investigation suggested that 4b might inhibit intracellular viral replication or the late stage of viral infection.
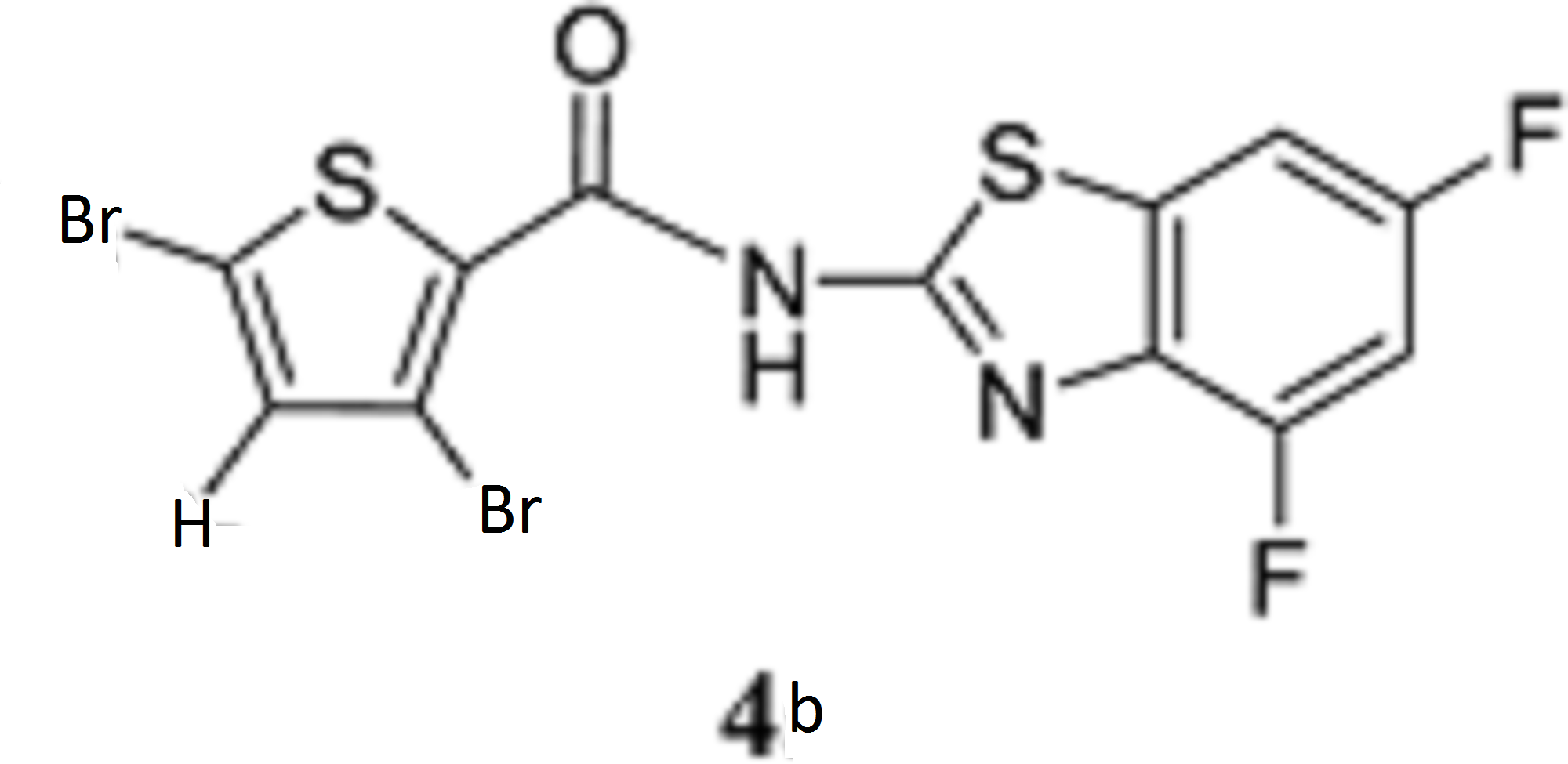
According to the same procedure used for 2f, starting from 3,5-dibromothiophene-2-carboxylic acid (286 mg, 1.00 mmol) and 4,6-difluorobenzo[d]thiazol-2-amine (204 mg, 1.10 mmol), 4b (270 mg, 60%) was obtained as white powder. mp: 245–246°C. 1H-NMR (DMSO-d6) δ: 7.43 (1H, dt, J=10.2, 2.0 Hz), 7.56 (1H, s), 7.83 (1H, dd, J=8.4, 2.0 Hz). 13C-NMR (DMSO-d6) δ: 102.2 (dd, J=28.0, 23.1 Hz), 104.7 (dd, J=26.4, 3.3 Hz), 114.3, 118.4, 131.4 (d, J=7.4 Hz), 134.3 (d, J=10.7 Hz), 134.9, 135.2, 152.7 (d, J=241.2, 20.7 Hz), 158.3 (dd, J=242.2, 10.7 Hz), 159.0, 159.7. HPLC purity: >99%, ESI-MS m/z 453 [M+H]+.
Antiviral Activity and Cytotoxicity of Tetra-halogenated Hybrid Compounds
 |
|||||
|---|---|---|---|---|---|
| Compound | R6 | R7 | R8 | EC50 (µM)a) | CC50 (µM)b) |
| 4a | Cl | H | H | 2.1 | >100 |
| 4b | Br | H | Br | 0.53 | >100 |
| 4c | Cl | H | Cl | 1.1 | >100 |
| 4d | Cl | Cl | H | 1.4 | 31 |
a) EC50 was evaluated by the CPE reduction assay. 280 TCID50/50 µL of MNV and a dilution series of each compound were incubated for 30 min. The mixture was exposed to RAW264.7 cells for 1 h (in duplicate). b) Cytotoxicity was evaluated by the WST-8 assay. RAW264.7 cells were treated with dilution series of each compound (in triplicate) for 72 h.

Three Methods:Killing Norovirus with Good HygieneKilling Norovirus in Your HomeTreating NorovirusCommunity Q&A
Norovirus is a contagious virus that affects many people each year. You can get norovirus through interaction with an infected person, by eating contaminated food, touching contaminated surfaces, or drinking contaminated water. However, there are ways to kill norovirus before it infects you. To do this, you will have to maintain personal hygiene and keep your home contamination-free.














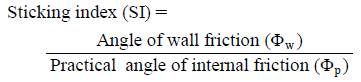 |
A larger SI indicates a greater likelihood that sticking will occur.
One cause of sticking is that when a powder is being compacted, the adhesive force between powder particles of the tablet and the punch surface exceeds the adhesive forces of powder particles within the tablet. Φp represents the frictional force acting between particles in the powder bed, and Φw represents the frictional force between the powder and the punch surface. The larger these values, the higher the friction and adhesion of the powder. We defined SI, which represents the degree of adhesion of a powder to the punch surface, as the value obtained by dividing Φw by Φp according to the following formula.
Sticking is a failure of pharmaceutical production that occurs when a powder containing a large amount of adhesive is being tableted. This is most frequently observed when long-term tableting is carried out, making it extremely difficult to predict its occurrence during the tablet formula design stage. The efficiency of the pharmaceutical production process could be improved if it were possible to predict whether a particular formulation was likely to stick during tableting. To address this issue, in the present study we prepared tablets composed of blended ibuprofen (Ibu), a highly adhesive drug, and measured the degree of adherence of powder particles to the surface of the tablet punch. We also measured the shear stress of the powder to determine the practical angle of internal friction (Φp) of the powder bed as well as the angle of wall friction (Φw) relative to the punch surface. These values were used to define a sticking index (SI), which showed a high correlation with the amount of Ibu that adhered to the punch during tableting; sticking occurred at SI >0.3. When the amount of lubricant added to the formulation was changed to yield tablets exhibiting different SI values without changing the compounding ratio, sticking did not occur at SI ≤0.3. These results suggest that determining the SI of a pharmaceutical powder before tableting allows prediction of the likelihood of sticking during tableting.
///////////sticking, shear stress, internal friction angle, wall friction angle, sticking index, ibuprofen, Tablet Production, Shear Testing, Pharmaceutical Powder
![]()
A Process Monitoring Solution that is Flexible and Scalable
The Unscrambler® X Process Pulse II can be utilised in all levels of an organisation, providing solutions to a variety of challenges using real-time process…
02 June 2016 by CAMO Software
The Unscrambler® X Process Pulse II can be utilised in all levels of an organisation, providing solutions to a variety of challenges using real-time process monitoring.
The device can be applied across many different industries and research fields to improve product development, manufacturing, and quality control, with powerful multivariate models.
Data analysts often face challenges with the data they want to analyse, which can be in different formats, coming from different systems, or it can be a mix of historical and live data and contain a large number of variables.
Unscrambler® X Process Pulse II handles all of these challenges, and translates the data into what is actually happening in the production process.
Download available at ………
/////////// Process Monitoring, Solution, Flexible, Scalable

Fig. 1. Schematic Representation of a Complex Fluidized-Bed Granulator
1: Exhaust air, 2: bag filter, 3: partition tube, 4: impeller, 5: rotor disc, 6: inlet air, 7: screen, 8: spray nozzle.
A new type of fluidized-bed granulator equipped with a particle-sizing mechanism was used for the preparation of fine particles that improved the solubility of a poorly water-soluble drug substance. Cefteram pivoxyl (CEF) was selected as a model drug substance, and its solution with a hydrophilic polymer, hydroxypropyl cellulose (HPC-L), was sprayed on granulation grade lactose monohydrate (Lac). Three types of treated particles were prepared under different conditions focused on the spraying air pressure and the amount of HPC-L. When the amount of HPC-L was changed, the size of the obtained particles was similar. However, particle size distribution was dependent on the amount of HPC-L. Its distribution became more homogenous with greater amounts of HPC-L, but the particle size distribution obtained by decreasing the spraying air pressure was not acceptable. By processing CEF with HPC-L using a complex fluidized-bed granulator equipped with a particle-sizing mechanism, the dissolution ratio was elevated by approximately 40% compared to that of unprocessed CEF. Moreover, in the dissolution profile of treated CEF, the initial burst was suppressed, and nearly zero order release was observed up to approximately 60% in the dissolution profile. This technique may represent a method with which to design fine particles of approximately 100 µm in size with a narrow distribution, which can improve the solubility of a drug substance with low solubility.
Three types of treated particles were prepared using a complex fluidized-bed granulator equipped with a particle-sizing mechanism under different conditions focused on the spraying air pressure and the amount of HPC-L. When the amount of HPC-L was changed, the size of the obtained particles was similar. However, particle size distribution was dependent on the amount of HPC-L. Its distribution became more homogenous with greater amounts of HPC-L, but the particle size distribution obtained by decreasing the spraying air pressure was not acceptable.
By processing CEF with HPC-L using this device, the dissolution ratio was elevated by approximately 40% compared to that of unprocessed one. Moreover, in the dissolution profile of treated CEF, the initial burst was suppressed, and nearly zero order release was observed up to approximately 60% in the dissolution profile.
The present method is applicable to the design of fine particles of approximately 100 µm in size with a narrow distribution, which improved the solubility of drug substance.
////////fine particle, particle size distribution, dissolution, complex fluidized-bed granulator
 |
|||
|---|---|---|---|
| Position | 1H-NMR (300 MHz, D2O) HOD=4.79 | 13C-NMR (75 MHz, D2O) DSSa)=−2.04 | HMBC correlation |
| 1 | — | 177.1 | C2-Ha, Hb |
| 2 | a: 2.49 (1H, dd, J=8.0, 16.0 Hz) | 40.9 | C3-H, C4-Ha |
| b: 2.63 (1H, dd, J=5.6, 16.0 Hz) | |||
| 3 | 5.63 (1H, m) | 67.5 | C2-Ha, Hb, C4-Hb |
| 4 | a: 3.60 (1H, d, J=14.0 Hz) | 68.9 | C2-Ha, Hb, C3-H, NMe |
| b: 3.86 (1H, dd, J=9.0, 14.0 Hz) | |||
| NMe3 | 3.18 (9H, s) | 54.5 | C4-Ha, Hb |
| 1′ | — | 175.6 | C2′-Ha, Hb |
| 2′ | a: 2.27 (1H, dd, J=7.0, 16.0 Hz) | 39.7 | C4′-Ha, C5′-Ha |
| b: 2.36 (1H, dd, J=7.4, 16.0 Hz) | |||
| 3′ | 0.98 (1H, m) | 6.7 | C4′-Ha, C5′-Ha, C2′-Ha, Hb |
| 4′ | a: 0.15 (1H, m) | 4.2 | C2′-Ha, Hb |
| b: 0.52 (1H, m) | |||
| 5′ | a: 0.15 (1H, m) | 4.4 | C2′-Ha, Hb |
| b: 0.52 (1H, m) | |||
a) DSS=sodium 2,2-dimethyl-2-silapentane-5-sulfonate.
To confirm the structure of 4, the identical carnitine ester was synthesized by condensation of (R)-carnitine and cyclopropylacetic acid using an acid chloride method (see Experimental). The 1H- and 13C-NMR data of the natural and synthetic samples were identical, and the absolute configuration was also determined to be R by comparing the specific rotation of the synthetic compound and that of the natural one. Thus, the isolated cyclopropane-containing new compound was confirmed to be cyclopropylacetyl-(R)-carnitine (4). Interestingly, carnitine esters, including a cyclopropylcarboxylic acid ester, were also isolated from a Boletaceae mushroom.9) We examined the toxicity of 4 in mouse by an intraperitoneal route; however, no toxicity was detected.
Cyclopropylacetyl-(R)-carnitine is specific to genuine R. subnigricans and sufficiently stable under ordinary experimental conditions. In addition, the upfield signals in the 1H-NMR spectrum corresponding to the cyclopropane core are easily recognizable in the 1H-NMR spectrum of crude mixtures of fruiting bodies; therefore, it would be a useful chemical marker for the identification of genuine R. subnigricans (Fig. 3).
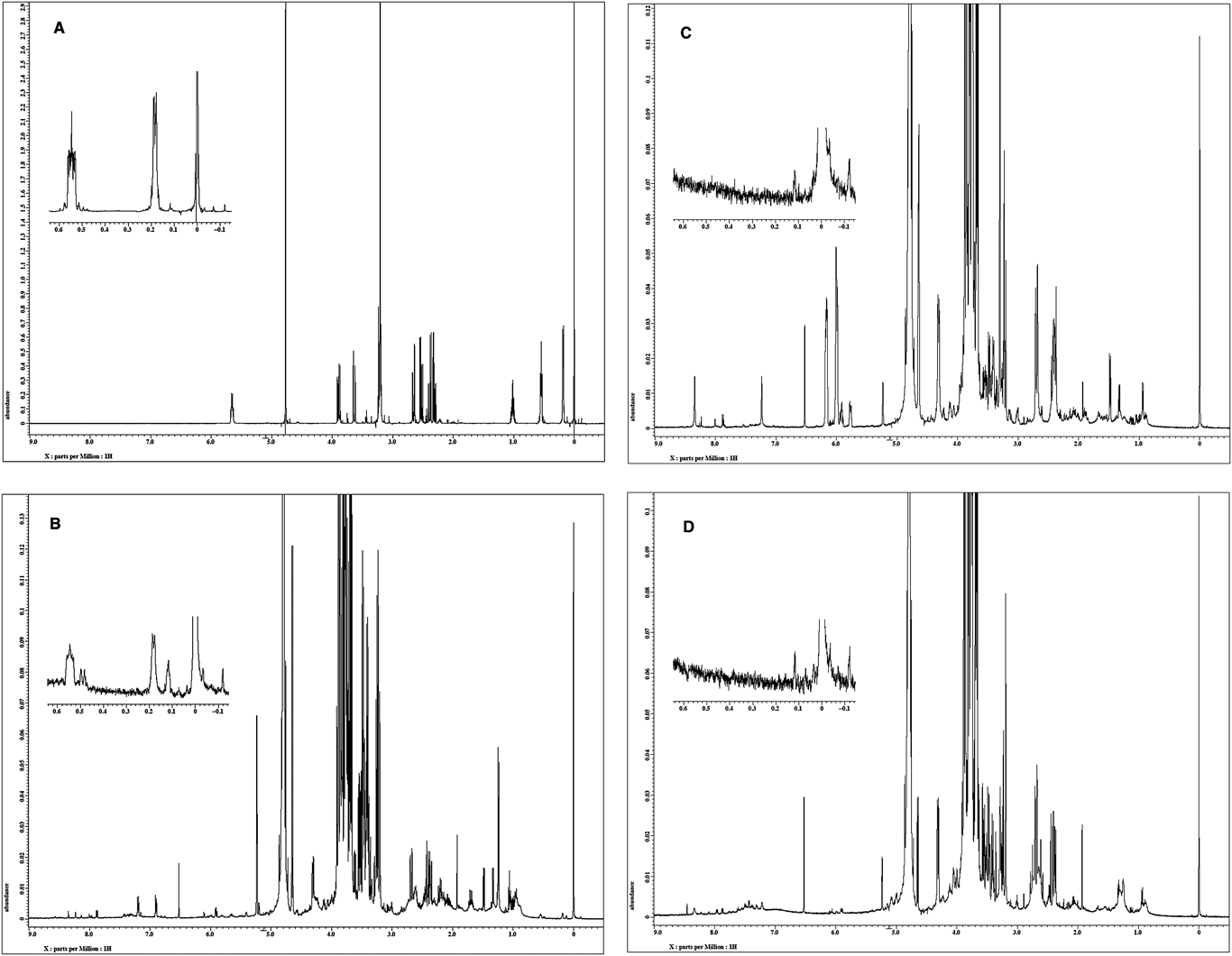 |
Fig. 3. 1H-NMR Spectra (500 MHz, D2O, TSPa)=0.00 ppm)(A) Authentic sample of cyclopropylacetyl-(R)-carnitine (4). (B) Crude water extract of R. subnigricans collected in Kyoto. (C) Crude water extract of Russula sp. collected in Miyagi. (D) Crude water extract of Russula sp. collected in Saitama. a) TSP=3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt. |
The fruiting bodies (500 g) of Russula subnigricans (collected in Kyoto) were cut into pieces and soaked in aqueous 0.3% AcOH (1.5 L) at 4°C overnight. The extract was filtered through filter paper under suction and then the filtrate was concentrated to about 100 mL under reduced pressure. The concentrated solution was dialyzed (relative molecular mass (Mr) 14000) against aqueous 0.3% AcOH (2.0 L×2) overnight. The dialyzate was concentrated to dryness and lyophilized to give a crude extract (27.1 g). A part (4.8 g) of the crude extract was dissolved in 1% AcOH in MeOH (48 mL), and then the soluble part was applied to an alumina column (aluminium oxide 90 standardized, Merck, 32 g), which was eluted with 1% AcOH in MeOH (300 mL). The eluate was concentrated to 5 mL, and this was diluted with aqueous 0.3% AcOH (10 mL) and chromatographed on ODS (Cosmosil 140C18 OPN, 16 g) which was eluted with H2O (300 mL) and 50% aqueous MeOH (100 mL). After removal of MeOH from the 50% MeOH fraction, the aqueous solution was washed with EtOAc (100 mL×3). The aqueous layer was concentrated in vacuo and then chromatographed on ODS by elution with 20% MeOH. The obtained fractions which contained a cyclopropane derivative were concentrated (16.2 mg) and purified by preparative TLC (ODS, 20% CH3CN) followed by HPLC (ODS ϕ6×250 mm, eluted with H2O for 10 min and then linear gradient from H2O to 20% CH3CN for 50 min at a flow rate of 1.5 mL/min with monitoring at 210 nm) to give 4 (3.4 mg, tR=20.9 min) as colorless syrup. Rf=0.31 (ODS, 1 : 4 MeOH–H2O); IR νmax (KBr): 3735, 3468, 1732, 1594, 1398, 1188, 1054 cm−1; LR-FAB-MS m/z 244 [M+H]+, 162 [M−C5H6O]+, HR-FAB-MS m/z 244.1572 [M+H]+; Calcd for C12H22NO4, 244.1549; [α]D31 −14.5 (H2O, c=0.96).
To a stirred cyclopropylacetic acid (0.098 mL, 1.05 mmol) was added thionyl chloride (0.080 mL, 1.10 mmol) and the mixture was stirred at room temperature (rt) for 1 h. To the crude acid chloride was directly added (R)-carnitine (86.0 mg, 0.533 mmol) and the mixture was stirred at rt for 1.5 h. After evaporation, the residue was dissolved into H2O and the aqueous layer was washed with EtOAc. The aqueous layer was applied to ODS column chromatography (H2O). The eluate was neutralized by saturated aqueous NaHCO3 solution and then concentrated to dryness. The resulting residue was extracted with MeOH and it was filtered through Celite. The filtrate and washings were concentrated in vacuo to afford cyclopropylacetyl-(R)-carnitine (4) (60.4 mg, 47% yield) as colorless foam. mp 180°C (decomp.);Rf=0.31 (ODS, 1 : 4 MeOH–H2O); IR νmax (KBr): 3735, 3433, 1734, 1592, 1392, 1179, 1034 cm−1; 1H-NMR (D2O, HOD=4.79) δ: 0.15 (2H, m, H-4′, 5′), 0.52 (2H, m, H-4′, 5′), 0.99 (1H, m, H-3′), 2.28, (1H, dd, J=7.0, 16.0 Hz, H-2′), 2.36 (1H, dd, J=7.4, 16.0 Hz, H-2′), 2.49 (1H, dd, J=8.0, 16.0 Hz, H-2), 2.63 (1H, dd, J=5.6, 16.0 Hz, H-2), 3.18 (9H, s, 4-N+Me3), 3.61 (1H, d, J=14.0 Hz, H-4), 3.86 (1H, dd, J=9.0, 14.0 Hz, H-4), 5.63 (1H, m, H-3); 13C-NMR (D2O, DSS=–2.04) δ: 4.2 (C4′, 5′), 4.4 (C4′, 5′), 6.7 (C3′), 39.7 (C2′), 40.9 (C2), 54.5 (4-N+Me3), 67.5 (C3), 68.9 (C4), 175.7 (C1′), 177.2 (C1); LR-FAB-MS m/z 244 [M+H]+, 162 [M−C5H6O]+, HR-FAB-MS m/z 244.1555 [M+H]+; Calcd for C12H22NO4, 244.1549; [α]D34 −16.6 (H2O, c=0.67).
A toxic mushroom, Russula subnigricans, causes fatal poisoning by mistaken ingestion. In spite of the potent bioactivity, the responsible toxin had not been identified for about 50 years since its first documentation. Recently, we isolated an unstable toxin and determined the structure. The slow elucidation was partly due to the instability of the toxin and also due to misidentification of R. subnigricans for similar mushrooms. To discriminate genuine Russula subnigricans from similar unidentified Russula species, we searched for a unique chemical marker contained in the mushroom. Cyclopropylacetyl-(R)-carnitine specific to R. subnigricans was identified as a novel compound whose1H-NMR signals appearing in the upfield region were easily recognizable among the complicated signals of the crude extract.
/////////cyclopropylacetyl-(R)-carnitine, cycloprop-2-ene carboxylic acid, russuphelin G, mushroom poisoning, Russula subnigricans,Russulaceae