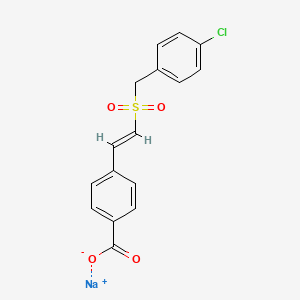
Racecadotril;
CAS 81110-73-8;
Acetorphan; Benzyl 2-(3-(acetylthio)-2-benzylpropanamido)acetate; Tiorfan; CADOTRIL;
benzyl 2-[[2-(acetylsulfanylmethyl)-3-phenylpropanoyl]amino]acetate
| Molecular Formula: | C21H23NO4S |
|---|---|
| Molecular Weight: | 385.47662 g/mol |

LAUNCHED 1993 BIOPROJET IN FRANCE
2010 IN CHINA
Racecadotril, also known as acetorphan, is an antidiarrheal drug which acts as a peripherally acting enkephalinase inhibitor.[2] Unlike other opioid medications used to treat diarrhea, which reduce intestinal motility, racecadotril has an antisecretory effect—it reduces the secretion of water and electrolytes into the intestine.[3] It is available in France (where it was first introduced in ~1990) and other European countries (including Germany, Italy, the UK and Spain) as well as most of South America and some South East Asian countries (including China, India and Thailand), but not in the United States. It is sold under the tradenames Hidrasec or, in France, Tiorfan.[4] In Italy it is sold under the tradename Tiorfix.[4]
Thiorphan is the active metabolite of racecadotril, which exerts the bulk of its inhibitory actions on enkephalinase.[5]
Racecadotril, also known as acetorphan, is an antidiarrheal drug which acts as a peripherally acting enkephalinase inhibitor. Unlike other medications used to treat diarrhea, which reduce intestinal motility, racecadotril has an antisecretory effect—it reduces the secretion of water and electrolytes into the intestine. It is available in India (It was first introduced in 1993 and is widely used in France) and other European countries, as well as most of South America and some South East Asian countries, but not in the United States. It is sold under the tradenames Hidrasec or, in France, Tiorfan. In Italy it is sold under the tradename Tiorfix.
In india trade names REDOTTIL, ZEDOTT .
A small randomized controlled trial found racecadotril to significantly reduce the duration and volume of watery diarrhea in children when given as an adjunct to oral rehydration therapy.
Racecadotril, also known as acetorphan, is an antidiarrheal drug which acts as a peripherally acting enkephalinase inhibitor. Unlike other medications used to treat diarrhea, which reduce intestinal motility, racecadotril has an antisecretory effect—it reduces the secretion of water and electrolytes into the intestine. It is available in India (It was first introduced in 1993 and is widely used in France) and other European countries, as well as most of South America and some South East Asian countries, but not in the United States. It is sold under the tradenames Hidrasec or, in France, Tiorfan. In Italy it is sold under the tradename Tiorfix.
In india trade names REDOTTIL, ZEDOTT .
A small randomized controlled trial found racecadotril to significantly reduce the duration and volume of watery diarrhea in children when given as an adjunct to oral rehydration therapy.
Indication & Dosage
Acute diarrhoea
Adult: 100 mg tid, up to 7 days.
Administration – May be taken with or without food.
Special Precautions – Renal insufficiency, pregnancy, lactation.
Adverse Drug Reactions – Vomiting, nausea, constipation, abdominal pain, thirst, vertigo and headache.
Mechanism of Action
Racecadotril increases the availability of endogenous opioids (enkephalins) by inhibiting the membrane-bound enkephalinase. These enkephalins activate δ-opioid receptors in the GI tract. This leads to a reduction in cAMP mucosal levels, resulting in a reducted secretion of water and electrolytes in the intestinal lumen. Onset: Within 30 min.

Foreign Names
Racecadotrilum (Latin)
Racecadotril (German)
Racécadotril (French)
Racecadotrilo (Spanish)
Brand Names
AD – Hetero, India
Aquasec – Micro Nova, India
Cadotril – Medifarma, Peru
Diarfix – CristerS, France
Dirasec – Abbott, India
Du La Bao – Baili Pharmaceutical, China
Enuff – Hetero, India
Feng Hai Ting – Zhengda Fenghai Pharmaceutical, China
Hidrasec – Abbott, China; Abbott, Philippines; Bagó, Ecuador; Ferrer, Costa Rica; Ferrer, Dominican Republic; Ferrer, Guatemala; Ferrer, Honduras; Ferrer, Mexico; Ferrer, Nicaragua; Ferrer, Panama; Ferrer, El Salvador; Fournier, Bulgaria; Fournier, Romania; Galenica, Greece; Laboratoire Sophartex, Vietnam; Leti, Venezuela; Silesia, Chile;
Solvay, Thailand
Hidrasec Children (pediatric) – Fournier, Bulgaria
Hidrasec Infants (pediatric)-Fournier, Bulgaria
Hydral – Alembic, India
Lomorest – Zuventus, India
Mo Ni Ka – Hailing, China
Mold – Aversi, Georgia
Pedidot – Elder, India
Racaril -East West, India
Racedot – Macleods, Georgia
Race-F – Bestochem, India
Racotil – Cipla, India
Racy – Abbott, India
Redotil – Dr. Reddy’s, India
Resorcal Lactantes/Ninos (pediatric) – Andromaco, Chile
Resorcal – Andromaco, Chile
Tiorfan – Abbott, Germany; Bago, Brazil; Bioproject, Tunisia; Bioprojet, France; CPH, Portugal; Ferrer, Peru; Ferrer Farma, Spain
Tiorfan Infantil – CPH, Portugal
Tiorfan Niños (pediatric) – Ferrer, Peru
Tiorfan nourrissons (pediatric) – Bioproject, Tunisia
Tiorfanor – Bioprojet, France
Tiorfast – Bioprojet, France
Tiorfix – Costanzafarma, Italy
Zomatril – FDC, India
See also
- Ecadotril, the (S)-enantiomer of racecadotril
Racecadotril (CAS NO.: 81110-73-8), with its systematic name of Glycine, N-(2-((acetylthio)methyl)-1-oxo-3-phenylpropyl)-, phenylmethyl ester, (+-)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: 2-Benzylacrylic acid (I) reacts with SOCl2 in hot toluene to afford the acyl chloride (II), which is condensed with N-tosylglycine benzyl ester (III) in the presence of TEA in toluene to yield the corresponding amide (IV). Finally, this compound is condensed with thioacetic acid by heating at 80 °C to afford the target acylthio compound.

Racecadotril is a neutral endopeptidase inhibitor used as antidiarrheal in the treatment of chronic cardiac insufficiency and is available under the brand names Hidrasec and Tiorfan. Racecadotril is chemically known as N-[2-[(acetylthio) methyl]- l-oxo-3-phenylpropyl] glycine phenyl methyl ester, (herein after referred by its generic name racecadotril) and represented by the formula (I).

U.S. Patent No. US 4,513,009 describes amino acid derivatives including racecadotril, a pharmaceutical composition and a method of treatment.
The US’009 patent also discloses a process for the preparation of racecadotril which is illustrated by below scheme:

U.S. Patent No. US 6,835,851 B2 discloses a process for the preparation of racecadotril which is illustrated by scheme below:

European Patent No. EP 0501870B 1 discloses a process for the preparation of racec

Racecadotril
The use of coupling agents like hydroxyl benzotriazole (HOBT) and dicyclohexyl amine carbodiimide (DCC) generally induces the formation of side products such as dicyclohexylurea. These side products do lead to major problems, wherein purification by chromatography may be contemplated, but the side products are extremely difficult to remove on an industrial scale.
Consequently, efforts have been made to replace the peptidic coupling step
so as to avoid the formation of side products associated with the use of the coupling agents. Thus, it appears that, even if the preparation of N-(mercaptoacyl)amino acid derivatives from .alpha.-substituted acrylic acids by Michael addition of a thio acid and conversion of acid to acid chloride by using thionyl chloride and then coupling of an amino ester may be advantageous on a laboratory scale, such reactions are difficult to adapt on an industrial use.
The aforementioned processes described above involves expensive reagents such as hydroxyl benzotriazole (HOBT) and dicyclohexyl amine carbodiimide (DCC) and hazardous reagent like thionyl chloride thus rendering the processes expensive and not feasible on industrial scale.
SYNTHESIS BY WORLDDRUGTRACKER

Patent
https://www.google.com/patents/WO2013098826A1?cl=en
EXAMPLES
Example-1: Preparation of Racecadotril (I):
Step A) Preparation of 2-acetyIsulfanyI methyI-3-phenyI propionic acid (IV)
16.2 g of 2-benzylacrylic acid and 12.3 ml of thioacetic acid were were charged into a clean and dry R.B.flask and stirred at about 30°C for about 1 hour. The reaction mixture was heated to about 60°C and stirred for about 4 hours.The excess of thioacetic acid was distilled off completely to afford the title compound as residue. Yield: 23.8 g. Step B) Preparation of Racecadotril crude (la)
23.8 g. of 2-acetyl sulfanylmethyl-3-phenyl-propionic acid (IV), 200ml of methylene chloride and 16.7 ml of triethylamine were charged into a clean and dry R.B.flask. 10. 5 ml of ethylchloroformate was added at about -5°C. The resultant reaction mixture was stirred at about 0°C for about 30 min. 33.7 g of glycine benzyl ester p-tosyalte (II), 14 ml of triethylamine and 100ml of methylene chloride was added as a mixture to the reaction mass at about 0°C. Then the resultant reaction mixture was stirred at about 0°C for about 1 hr. followed by at about 30°C for about 30 min. After completion of the reaction as determined by TLC, the reaction mass was washed with 65 ml of distilled water, 65 ml 4% sodium bicarbonate solution and followed by 65 ml distilled water. The organic and aqueous phases were separated and the solvent was distilled completely, 2 x 50 ml Isopropyl alcohol was charged and again distilled off the solvent completely to give residue. The residue
obtained was triturated with a mixture of isopropyl alcohol 4 ml) and n-hexane (94 ml) at about 5°C to the title compound as crude. Yield: 34 g.
ExampIe-2: Purification of Racecadotril (Crude):
34 g. of crude Racecadotril and 35 ml of 20 % v/v aqueous .methanol were charged in a clean and dry R.B.flask and heated to about 65°C. 3g. of SP.carbon was charged and stirred at about 65°C for about 10 min. The reaction suspension was stirred at about 65°C for about 10 min. The reaction suspension was filtered on hyflow bed (diatomous earth) and washed the hyflow bed with 30 ml of aqueous methanol. The filtrate obtained was cooled to about 0°C for about 30 min. The solid separated was filtered and the solid obtained washed with 60 ml of precooled aqueous methanol to afford the pure racecadotril (I).
Yield: 29 g.; Purity by HPLC: 99.5 area %; The overall yield is 75.3%.
PATENT
https://www.google.com/patents/CN104356036A?cl=en
Example 1
The 40. 0g Racecadotril dissolved in 200ml of absolute ethanol and water bath heated to 40 ° C, and stir until the whole solution, stirring was stopped, the solution was placed in 15 ° C water bath was allowed to stand, when starting When there is precipitation of crystals, and then placed under the 0 ° C crystallization, after filtration, to 45 ° C under hot air drying cycle 6 hours to obtain 29. 2g, purity 99.6% of Racecadotril a polymorph crystals.
reflection angle X-ray powder diffraction pattern 20 at 4.3 °, 8.7 °, 13.2 °, 16.8 °, 17.8 ° and 20.0 ° at the show X-ray powder diffraction peaks. In 1135. 19CHT1,1551. 46CHT1,1644. 73CHT1,1687. 57CHT1, 1731. 35CHT1 and 3289. 20CHT1 displayed at an infrared absorption peak.
Clips

US 20020055645

PATENT
Racecadotril, chemical name N_ [(R, S) -3- acetyl-mercapto-2-benzyl-propionyl)] glycine benzyl ester, is a neprilysin inhibitor, selectively, reversible inhibition of neprilysin, so that the inner protection from degradation of endogenous enkephalins, prolong the physiological activity of endogenous enkephalins in the digestive tract, mainly used in clinical treatment of children and adults with acute diarrhea. Its structural formula is as follows:

Racecadotril as enkephalinase inhibitors, developed in France in 1993 Bioprojet listed acute diarrhea treatment, trade name Tiorfan.
In W02011116490A1, US5945548 and CN101768095A and other documents, documented racecadotril the synthesis process, but did not report the crystal form; therefore the present inventors have not reported Racecadotril crystalline polymorph conduct further
Example 1
[0032] The 40. 0g Racecadotril dissolved in 200ml of absolute ethanol and water bath heated to 40 ° C, and stir until the whole solution, stirring was stopped, the solution was placed in 15 ° C water bath was allowed to stand, when starting When there is precipitation of crystals, and then placed under the 0 ° C crystallization, after filtration, to 45 ° C under hot air drying cycle 6 hours to obtain 29. 2g, purity 99.6% of Racecadotril a polymorph crystals.
[0033] reflection angle X-ray powder diffraction pattern 20 at 4.3 °, 8.7 °, 13.2 °, 16.8 °, 17.8 ° and 20.0 ° at the show X-ray powder diffraction peaks. In 1135. 19CHT1,1551. 46CHT1,1644. 73CHT1,1687. 57CHT1, 1731. 35CHT1 and 3289. 20CHT1 displayed at an infrared absorption peak.
research.
References
- 1 “SPC-DOC_PL 39418-0003.PDF” (PDF). Medicines and Healthcare Products Regulatory Agency. Bioprojet Europe Ltd. 26 December 2012. Retrieved 7 May 2014.
- 2Matheson AJ, Noble S (April 2000). “Racecadotril”. Drugs 59 (4): 829–35; discussion 836–7. doi:10.2165/00003495-200059040-00010. PMID 10804038.
- 3Matheson, AJ; Noble, S (April 2000). “Racecadotril.”. Drugs 59 (4): 829–35; discussion 836–7. doi:10.2165/00003495-200059040-00010. PMID 10804038.
- 4Brayfield, A, ed. (13 December 2013). “Racecadotril”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 6 May 2014.
- 5Spillantini MG, Geppetti P, Fanciullacci M, Michelacci S, Lecomte JM, Sicuteri F (June 1986). “In vivo ‘enkephalinase’ inhibition by acetorphan in human plasma and CSF”. European Journal of Pharmacology 125 (1): 147–50. doi:10.1016/0014-2999(86)90094-4. PMID 3015640.
- 6 http://www.ingentaconnect.com/content/govi/pharmaz/2006/00000061/00000012/art00005?crawler=true

| CN101103960A * | Jul 14, 2006 | Jan 16, 2008 | 海南盛科生命科学研究院 | Dry mixed suspension containing racecadotril and preparation method thereof |
| CN101768095A * | Dec 26, 2008 | Jul 7, 2010 | 山东齐都药业有限公司 | Preparation method of racecadotril |
| WO2001097803A1 * | Jun 20, 2001 | Dec 27, 2001 | Laboratoire Glaxosmithkline | Pharmaceutical preparations comprising racecadotril (acetorphan) |
| WO2013098826A1 * | Dec 26, 2011 | Jul 4, 2013 | Symed Labs Limited | “a process for the preparation of n-[2-[(acetylthio) methyl]-1-oxo-3-phenylpropyl] glycine phenyl methyl ester and intermediates thereof” |
| Reference |
|---|
| CN101103960A * | Jul 14, 2006 | Jan 16, 2008 | 海南盛科生命科学研究院 | Dry mixed suspension containing racecadotril and preparation method thereof |
| CN101768095A * | Dec 26, 2008 | Jul 7, 2010 | 山东齐都药业有限公司 | Preparation method of racecadotril |
| WO2001097803A1 * | Jun 20, 2001 | Dec 27, 2001 | Laboratoire Glaxosmithkline | Pharmaceutical preparations comprising racecadotril (acetorphan) |
| WO2013098826A1 * | Dec 26, 2011 | Jul 4, 2013 | Symed Labs Limited | “a process for the preparation of n-[2-[(acetylthio) methyl]-1-oxo-3-phenylpropyl] glycine phenyl methyl ester and intermediates thereof” |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | 金庆平 等: “神经内肽酶抑制剂消旋卡多曲(Racecadotril)的合成工艺研究“, 《中国现代应用药学杂志》, vol. 20, no. 7, 31 August 2003 (2003-08-31) | ||
| US6013829 * | Feb 4, 1997 | Jan 11, 2000 | Societe Civile Bioprojet | Process for the asymmetric synthesis of S-acyl derivatives of 2-mercaptomethyl -3- phenyl propanoic acid, application to the synthesis of N-(mercaptoacyl) amino acid derivatives |
| US20040009956 * | Apr 29, 2003 | Jan 15, 2004 | Dehua Pei | Inhibition of protein tyrosine phosphatases and SH2 domains by a neutral phosphotyrosine mimetic |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | MOHAMED A.O. ET AL.: ‘Stability-indicating methods for the determination of racecadotril in the presence of its degradation products‘ BIOSCIENCE TRENDS vol. 3, no. 6, 2009, pages 247 – 252, XP055074337 | ||
| Citing Patent | Filing date | Publication date | Applicant | Title |
| CN104356036A * | Nov 7, 2014 | Feb 18, 2015 | 山东齐都药业有限公司 | Alpha crystal form of racecadotril and preparation method of alpha crystal form |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(RS)-Benzyl N-[3-(acetylthio)-2-benzylpropanoyl]glycinate
|
|
| Clinical data | |
| Trade names | Hidrasec, Tiorfan |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 90% (active thiorphan metabolite)[1] |
| Metabolism | Liver-mediated[1] |
| Biological half-life | 3 hours[1] |
| Excretion | Urine (81.4%), feces (8%)[1] |
| Identifiers | |
| CAS Number | 81110-73-8 |
| ATC code | A07XA04 (WHO) |
| PubChem | CID 107751 |
| ChemSpider | 96913 |
| UNII | 76K53XP4TO |
| ChEMBL | CHEMBL2103772 |
| Synonyms | Benzyl 2-[3-(acetylthio)-2-benzylpropanamido]acetate |
| Chemical data | |
| Formula | C21H23NO4S |
| Molar mass | 385.47662 g/mol |
| Chirality | Racemic mixture |
/////////Racecadotril, acetorphan, antidiarrheal drug
CC(=O)SCC(CC1=CC=CC=C1)C(=O)NCC(=O)OCC2=CC=CC=C2




















 Integrifolin
Integrifolin






 Journal title :
Journal title : 






























 Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.
Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company.