Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
How are you? Please allow me to introduce Pharmacodia global drug Database & services to you briefly.
Pharmacodia Global Pharmaceutical Intelligence Platform is the first tier drug database for R&D Professionals including 40,000+ Drug data, 13,817,000+ Patents, 488,000+ Registration & Approval, 414,000+ Clinical Trials and 63,000+ items related regulatory policies.
Focusing on the drug research and development, Pharmacodia global database includes all small molecule drugs and biologics since 1982 (either in developing or marketed in the world). You may have a three days free trial for the Database.
Website: https://data.pharmacodiaglobal.com
If your colleagues and friends like to have a trial for the database, would you please forward the email to them, they can easily register an account and apply for a one day free trail. Thank you very much.
SCROLL UP OR DOWN
Sincerely yours,
Jim
Chair of Pharmacodia Group
HongKong
If you are interested in knowing more about our database and services, please contact us through email/whatsApp:
The “e Manufacturing Execution System” for pharmaceutical manufacturing is different from eMES in any other manufacturing industry. The difference primarily comes from the stringent Regulatory Compliance norms to be adhered to in Pharmaceutical Operations. In the Pharmaceutical Manufacturing the eMES is also referred as eBMR or “e Batch Manufacturing Record”.
The key objectives of implementing “e Manufacturing Execution Systems” (or eBMR) in Pharma Manufacturing are:
Achieve Compliancewith global regulatory norms, quality processes and ALCOA + norms.
Achieve Data integrityacross functions, systems and equipment records.
Audit readiness: Become ever ready for audit, with complete electronic records.
Paperless operation: Achieve paperless (or less paper) operation at the plant.
Pharmaceutical companies operate in one of the most dynamic and highly regulated environments. Regulatory audit frequencies have increased. Warning letters & import restrictions incidences have increased alongside. Many companies fail in clearing Regulatory Audits. Failing an audit results in over two years of business loss and huge expenses in achieving success with re-audit.
Most of the Pharmaceutical Manufacturing organizations have realized that they must implement a comprehensive eMES/eBMR systems for achieving the stated objective for long term business survival in todays regulated environment. However, the existing infrastructure scenario, inhibit moving forward with such implementation.
The scenario: Many Pharmaceutical Manufacturing Units are supplying to strongly regulated markets. They have a mix of the following infrastructure scenario:
Obsolete Equipment: Some equipment are obsolete in their design. These have no digital ability to record and communicate critical operations parameters for recording into BMR.
Semi-modern equipment: Some equipment have some digital ability to show records but are not enabled for automatic data transmission to SCADA database.
Modern Equipment: Some equipment are modern, PLC controlled and they can store digital data for some time. They still need to be integrated to SCADA (Supervisory Control And Data Acquisition) system.
There are also several cases of failures in implementing eMES solution provided by global brands providing the MES solution. Some of the key reasons of eMES implementation project failures are:
Complexity of the eMESSoftware systems. The level of complexity that can be digested across continents vary a lot. What is simple for some European countries may be too complex for Asian countries.
Design and Architecture of MES Solutions: A generic MES, modified for Pharma, would not succeed in Pharma. The eMES must be designed for Pharmaceutical operations, where the regulatory compliance requires flexibility in system to allow special process steps, for which there is no global best practices. The system must be architected for allowing new recipe, new product and new process to be quickly configured.
Right collaboration: Very often IT Team of Pharma companies would be good at handling the ERP Software and Level 3 & Level 4 IT systems. They would lack expertise of Level 1 and Level 2 Automation. Companies need to get right IT Solution providers capable of supplying and implementation right eMES along with Automation Solutions.
Comprehensive Solution: A comprehensive solution design would encompass eMES, integration with ERP, integration with SCADA and direct integration with many devices. The SCADA and Automation solution would include upgrading the semi modern equipment, connecting them along with modern equipment to SCADA. Adopting right strategy solution for obsolete equipment.
Phased implementation Strategy: There is variety of strategies for eMES implementation with complete automation integration. Designing and selecting right strategy is a Critical Success Factor.
The country, the facility location, equipment vendors, employees, organization culture, the product and market combination etc., makes every unit and unique unit. The eMES solution must be designed to suit each such unit. One solution would not fit all. The solutions must be configurable for taking care of the uniqueness. The solutions must be robust and maintainable.
Laurel MES™ product suite for “e Manufacturing Execution” is architected and designed for Pharmaceutical Manufacturing businesses to achieve compliance and audit readiness. Laurel MES™ comes along with Automation Solution and Systems Integration Solution which provides a complete, comprehensive and configurable solution. The drug product variants, market variants and product changeover challenges are addressed as required. Management dashboards and scorecards are fascinating. This solution is integration-ready for latest manufacturing and testing equipment.
Experienced Chief Executive Officer with a track record of leading Architecture Design and development of Software Products for Pharma Manufacturing Execution, Pharma R&D and making it successful in the market. Demonstrated strength in the information technology, software technologies, software services, Pharma, Chemical and Engineering industry. Skilled in Business Process, Architecture and Design of Enterprise Software and Computer Integrated Manufacturing. Strong business development professional with Engineering and MBA qualifications.
Welcome to the PharmaCamp with Neha. With this video channel. I would like to spread knowledge about the pharmaceutical world. This is a small initiative from my side to share knowledge with the world, as I feel education is the best gift one can receive or give back to society. Are you a working professional who wants to upgrade your skills? OR a student who wants to make a career in pharmaceuticals? OR are you a person who is interested to learn about the regulatory systems? Then this YouTube channel is for you. In this video, we will about US FDA Investigational New Drug (IND) Application. In my upcoming video, I will provide the stepwise approach for these IND applications. Therefore, this video will serve as the basis for those upcoming videos to explain the concept of INDs.
About me: I am Neha Parashar, working as a Senior Manager in a pharmaceutical company and based in Germany. I am a passionate healthcare professional, an educator, and a mentor. LinkedIn: http://linkedin.com/in/neha-parashar
An Active Substance Master File (ASMF), or formerly known as European Drug Master File (EDMF) procedure, is a set of documents that protects the valuable confidential intellectual property of the manufacturer.
The main objective of the Active Substance Master File (ASMF) procedure, formerly known as the European Drug Master File (EDMF) procedure, is to allow valuable confidential intellectual property or ‘know-how’ of the manufacturer of the active substance (ASM) to be protected, while at the same time allowing the Applicant or Marketing Authorisation (MA) holder to take full responsibility for the medicinal product and the quality and quality control of the active substance. National Competent Authorities/EMA thus have access to the complete information that is necessary for an evaluation of the suitability of the use of the active substance in the medicinal product.
Final Guideline on Active Substance Master File Procedure – Revision 4 (PDF/358.28 KB)
The ASMF holder may have an ASMF as well as a Certificate of Suitability (CEP) issued by EDQM for a single active substance. Generally, it is however not acceptable that the Applicant/MA holder refers to an ASMF as well as to a CEP for a single active substance of a particular MAA/MAV.
What is an ASMF in pharmaceutical industry?
The main objective of the Active Substance Master File (ASMF) procedure, formerly known as the European Drug Master File (EDMF) procedure, is to allow valuable confidential intellectual property or ‘know-how’ of the manufacturer of the active substance (ASM) to be protected, while at the same time allowing the …
What is the difference between Asmf and DMF?
An Active Substance Master File (ASMF) is the currently recognised term in Europe, formerly known as European Drug Master File (EDMF) or a US-Drug Master file (US-DMF) in the United States.
What is a EDQM CEP certificate?
To obtain a Certificate of Suitability to the monographs of the European Pharmacopoeia (CEP), applicants must send in electronic format the following documentation to the Certification of Substances Department (DCEP) of the EDQM: a completed application form which includes your invoicing details.
Electronic Active Substance Master Files (eASMF)
ASMF Holders who are supplying substances to more than one Centrally Authorised Product (CAP) should submit their ASMF to the Agency once and not for each application.
The use of eCTD is mandatory for all for centralised procedure human ASMF submissions since 1 July 2016. The use of eCTD is mandatory for ASMFs used for DCP/MRP human procedures from 1 January 2018. Use of eCTD is mandatory for new national MAAs since 1 July 2018 and from 1 January 2019 for all other submission types. The relevant guidance should be followed and the technical eCTD validation criteria must be passed.
To ensure that the above is followed promptly, please refer to the human EMA Pre-authorisation guidance carefully and apply for an EMEA/ASMF/XXXXX number at your earliest convenience by submitting the EMEA/ASMF request form.
For veterinary medicines the accepted electronic format is VNeeS and NeeS and for ASMFs also exceptionally eCTD is allowed. More information can be found from the Veterinary Pre-submission guidance.
It is very important to note that the above two initiatives are different.
The EMA eASMF submission rules are mandatory since 1st September 2013 and are applicable to all Centralised applications.
The EU Assessment worksharing initiative is valid for Centralised and Decentralised procedures.
A valid ASMF should have either an EMEA/ASMF number or an EU/ASMF number, depending on the intended use of the ASMF by its holder. When applying for EMEA or EU ASMF numbers, or submitting any documentation quoting these, please note that they are not inter-changeable. Only one ASMF number should be quoted.
It is mandatory to use XML delivery files for ASMF submissions using the eSubmission Gateway and the Web Client.
Letter of Access Template (Previously Annex 2), Template Administrative Information – (Previously Annex 3), Template Withdrawal of Access Letter –Should be filled in in accordance of the ASMF type (EU/ASMF/XXXXX or EMEA/ASMF/XXXXX)
ASMF worksharing – Introduction to the procedure (October 2018)
Requesting an EU_ASMF repository number (October 2018)
Submitting an initial worksharing ASMF (October 2018)
Determining the parent procedure (October 2018)
Assessment report template (October 2018)
Sharing assessment reports (October 2018)
Submitting a variation to an ASMF (October 2018)
Use of an approved ASMF in a new procedure (October 2018)
An Active Substance Master File (ASMF), or formerly known as European Drug Master File (EDMF) procedure, is a set of documents that protects the valuable confidential intellectual property of the manufacturer. The purpose of an ASMF is to provide the Health Authorities (HAs) with complete information of the active substance used in a drug product to evaluate its suitability for the product. It also allows the Marketing Authorization Holder (MAH) of the product to take the responsibility for the quality of the active substance used in the product.
ASMF Content
An ASMF must contain all the scientific information related to the active substance. The information of an ASMF is divided into two parts:
Applicant Part (AP) – Contains information considered as non-confidential by the MAH
Restricted Part – Contains confidential information
ASMF Usage
An ASMF is submitted only to support a Marketing Authorisation Application (MAA) or Marketing Authorisation Variation (MAV). It is used for the following active substances:
Active substances that are new
Already existing active substances, which are excluded from the European Pharmacopoeia (Ph. Eur.)
Pharmacopoeial active substances, which are included in the (Ph. Eur.)
An ASMF holder is required to submit the following to the MAH holder:
A copy of AP’s latest version
A copy of the summary/detailed version of AP’s latest version
A copy of the letter of access, which has not been previously submitted for the same product
A single active substance can have both, an ASMF as well as a Certificate of Suitability (CEP). However, the MAH cannot refer to both the documents for a single active substance. In case, the information present in a CEP is insufficient, only then both the documents can be referred.
Follow the journey of a medicine for human use assessed by EMA in this interactive timeline. It explains all stages from initial research to patient access, including how EMA supports medicine development, assesses the benefits and risks and monitors the safety of medicine
From laboratory to patient: the journey of a centrally authorised medicine (PDF/1.75 MB)
First published: 04/03/2019
Last updated: 10/02/2020
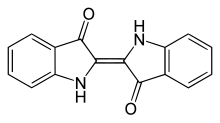
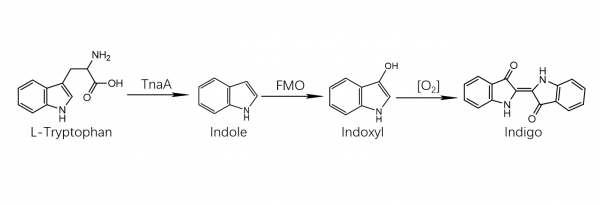
Indigo dye is an organic compound with a distinctive blue color. Historically, indigo was a natural dye extracted from the leaves of some plants of the Indigofera genus, in particular Indigofera tinctoria; dye wielding Indigofera plants were commonly grown and used throughout the world, in Asia in particular, as an important crop, with the production of indigo dyestuff economically important due to the previous rarity of some blue dyestuffs historically.[1]
Most indigo dye produced today is synthetic, constituting several thousand tons each year. It is most commonly associated with the production of denim cloth and blue jeans, where its properties allow for effects such as stone washing and acid washing to be applied quickly.
Uses
Indigo dye
The primary use for indigo is as a dye for cotton yarn, mainly used in the production of denim cloth suitable for blue jeans; on average, a pair of blue jeans requires just 3 grams (0.11 oz) – 12 grams (0.42 oz) of dye to produce. Smaller quantities are used in the dyeing of wool and silk.
Indigo carmine, also known as indigo, is an indigo derivative which is also used as a colorant. About 20 thousand tons are produced annually, again mainly for the production of blue jeans.[1] It is also used as a food colorant, and is listed in the United States as FD&C Blue No. 2.
A variety of plants have provided indigo throughout history, but most natural indigo was obtained from those in the genus Indigofera, which are native to the tropics, notably the Indian subcontinent. The primary commercial indigo species in Asia was true indigo (Indigofera tinctoria, also known as I. sumatrana). A common alternative used in the relatively colder subtropical locations such as Japan’s Ryukyu Islands and Taiwan is Strobilanthes cusia.
Until the introduction of Indigofera species from the south, Polygonum tinctorum (Dyer’s knotweed) was the most important blue dyestuff in East Asia; however, the crop produced less dyestuff than the average crop of indigo, and was quickly surpassed in terms of favour for the more economical Indigofera tinctoria plant. In Central and South America, the species grown is Indigofera suffruticosa, also known as anil. In Europe, Isatis tinctoria, commonly known as woad, was used for dyeing fabrics blue, containing the same dyeing compounds as indigo, also referred to as indigo.
Several plants contain indigo, which, when exposed to an oxidising source such as atmospheric oxygen, reacts to produce indigo dye; however, the relatively low concentrations of indigo in these plants make them difficult to work with, with the color more easily tainted by other dye substances also present in these plants, typically leading to a greenish tinge.
The precursor to indigo is indican, a colorless, water-soluble derivative of the amino acid tryptophan. Indican readily hydrolyzes to release β-D-glucose and indoxyl. Oxidation by exposure to air converts indoxyl to indigotin, the insoluble blue chemical that is the endpoint of indigo dye. Indican was obtained from the processing of the plant’s leaves, which contain as much as 0.2–0.8% of this compound. The leaves were soaked in water and fermented to convert the glycoside indican present in the plant to the blue dye indigotin.[2] They precipitate from the fermented leaf solution when mixed with a strong base[3] such as lye, pressed into cakes, dried, and powdered. The powder was then mixed with various other substances to produce different shades of blue and purple.
Natural sources of indigo also include mollusks; the Murex sea snail produces a mixture of indigo and 6,6′-dibromoindigo (red), which together produce a range of purple hues known as Tyrian purple. Light exposure during part of the dyeing process can convert the dibromoindigo into indigo, resulting in blue hues known as royal blue, hyacinth purple, or tekhelet.
Chemical synthesis
Heumann’s synthesis of indigo
Pfleger’s synthesis of indigo
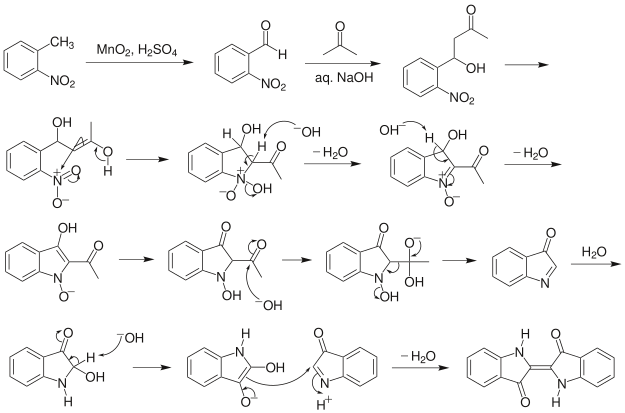
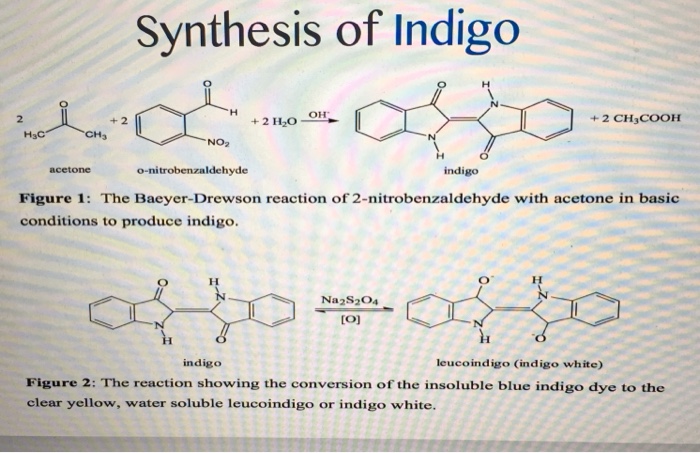
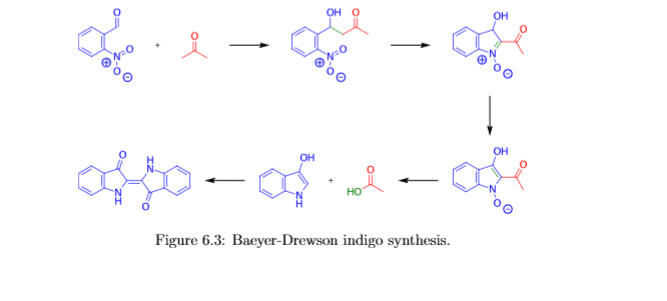
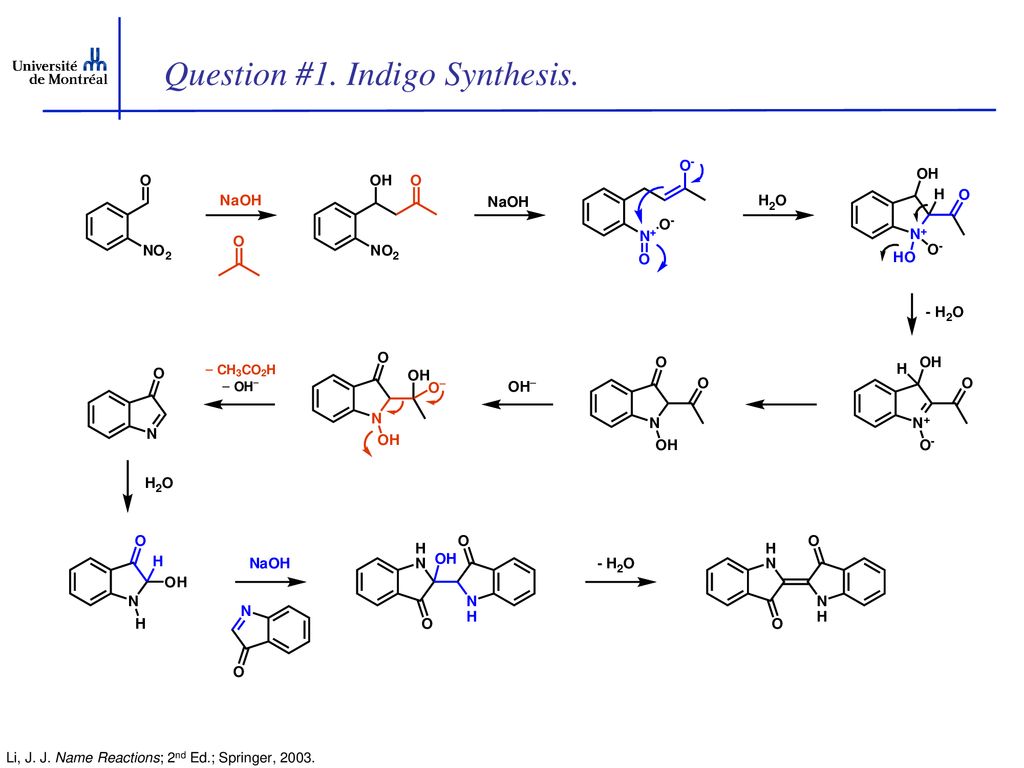
Given its economic importance, indigo has been prepared by many methods. The Baeyer-Drewson indigo synthesis dates back to 1882. It involves an aldol condensation of o-nitrobenzaldehyde with acetone, followed by cyclization and oxidative dimerization to indigo. This route is highly useful for obtaining indigo and many of its derivatives on the laboratory scale, but proved impractical for industrial-scale synthesis. Johannes Pfleger[4] and Karl Heumann (de) eventually came up with industrial mass production synthesis.[5]
The first commercially practical route of producing indigo is credited to Pfleger in 1901. In this process, N-phenylglycine is treated with a molten mixture of sodium hydroxide, potassium hydroxide, and sodamide. This highly sensitive melt produces indoxyl, which is subsequently oxidized in air to form indigo. Variations of this method are still in use today. An alternative and also viable route to indigo is credited to Heumann in 1897. It involves heating N-(2-carboxyphenyl)glycine to 200 °C (392 °F) in an inert atmosphere with sodium hydroxide. The process is easier than the Pfleger method, but the precursors are more expensive. Indoxyl-2-carboxylic acid is generated. This material readily decarboxylates to give indoxyl, which oxidizes in air to form indigo.[1] The preparation of indigo dye is practised in college laboratory classes according to the original Baeyer-Drewsen route.[6]
The oldest known fabric dyed indigo, dated to 6,000 years ago, was discovered in Huaca Prieta, Peru.[7] Many Asian countries, such as India, Japan, and Southeast Asian nations have used indigo as a dye (particularly silk dye) for centuries. The dye was also known to ancient civilizations in Mesopotamia, Egypt, Britain, Mesoamerica, Peru, Iran, and West Africa. Indigo was also cultivated in India, which was also the earliest major center for its production and processing.[8] The I. tinctoria species was domesticated in India.[8] Indigo, used as a dye, made its way to the Greeks and the Romans, where it was valued as a luxury product.[8]
India was a primary supplier of indigo to Europe as early as the Greco-Roman era. The association of India with indigo is reflected in the Greek word for the dye, indikón (Ἰνδικόν, Indian).[9] The Romans latinized the term to indicum, which passed into Italian dialect and eventually into English as the word indigo.
Cake of indigo, about 2 cm
In Mesopotamia, a neo-Babylonian cuneiform tablet of the seventh century BC gives a recipe for the dyeing of wool, where lapis-colored wool (uqnatu) is produced by repeated immersion and airing of the cloth.[9] Indigo was most probably imported from India. The Romans used indigo as a pigment for painting and for medicinal and cosmetic purposes. It was a luxury item imported to the Mediterranean from India by Arab merchants.
Indigo remained a rare commodity in Europe throughout the Middle Ages. A chemically identical dye derived from the woad plant (Isatis tinctoria) was used instead. In the late 15th century, the Portuguese explorer Vasco da Gama discovered a sea route to India. This led to the establishment of direct trade with India, the Spice Islands, China, and Japan. Importers could now avoid the heavy duties imposed by Persian, Levantine, and Greek middlemen and the lengthy and dangerous land routes which had previously been used. Consequently, the importation and use of indigo in Europe rose significantly. Much European indigo from Asia arrived through ports in Portugal, the Netherlands, and England. Many indigo plantations were established by European powers in tropical climates. Spain imported the dye from its colonies in Central and South America, and it was a major crop in Haiti and Jamaica, with much or all of the labor performed by enslaved Africans and African Americans. In the Spanish colonial era, intensive production of indigo for the world market in the region of modern El Salvador entailed such unhealthy conditions that the local indigenous population, forced to labor in pestilential conditions, was decimated.[10] Indigo plantations also thrived in the Virgin Islands. However, France and Germany outlawed imported indigo in the 16th century to protect the local woad dye industry.
Indigo was the foundation of centuries-old textile traditions throughout West Africa. From the Tuareg nomads of the Sahara to Cameroon, clothes dyed with indigo signified wealth. Women dyed the cloth in most areas, with the Yoruba of Nigeria and the Mandinka of Mali particularly well known for their expertise. Among the Hausa male dyers, working at communal dye pits was the basis of the wealth of the ancient city of Kano, and they can still be seen plying their trade today at the same pits.[11]
In Japan, indigo became especially important during the Edo period. This was due to a growing textiles industry,[12] and because commoners had been banned from wearing silk,[13] leading to the increasing cultivation of cotton, and consequently indigo – one of the few substances that could dye it.[14]
Newton used “indigo” to describe one of the two new primary colors he added to the five he had originally named, in his revised account of the rainbow in Lectiones Opticae of 1675.[15]
In North America indigo was introduced into colonial South Carolina by Eliza Lucas, where it became the colony’s second-most important cash crop (after rice).[16] As a major export crop, indigo supported plantation slavery there.[17] In the May and June 1755 issues of The Gentleman’s Magazine there appeared a detailed account of the cultivation of indigo, accompanied by drawings of necessary equipment and a prospective budget for starting such an operation, authored by South Carolina planter Charles Woodmason. It later appeared as a book. [18][19] By 1775, indigo production in South Carolina exceeded 1,222,000 pounds. [20] When Benjamin Franklin sailed to France in November 1776 to enlist France’s support for the American Revolutionary War, 35 barrels of indigo were on board the Reprisal, the sale of which would help fund the war effort.[21] In colonial North America, three commercially important species are found: the native I. caroliniana, and the introduced I. tinctoria and I. suffruticosa.[22]
Because of its high value as a trading commodity, indigo was often referred to as blue gold.[23]
The demand for indigo in the 19th century is indicated by the fact that in 1897, 7,000 km2 (2,700 sq mi) were dedicated to the cultivation of indican-producing plants, mainly in India. By comparison, the country of Luxembourg is 2,586 km2 (998 sq mi).[1]
In 1865 the German chemist Adolf von Baeyer began working on the synthesis of indigo. He described his first synthesis of indigo in 1878 (from isatin) and a second synthesis in 1880 (from 2-nitrobenzaldehyde). (It was not until 1883 that Baeyer finally determined the structure of indigo.[24]) The synthesis of indigo remained impractical, so the search for alternative starting materials at Badische Anilin- und Soda-Fabrik (BASF) and Hoechst continued. Johannes Pfleger[4] and Karl Heumann eventually came up with industrial mass production synthesis.[5]
The synthesis of N-(2-carboxyphenyl)glycine from the easy to obtain aniline provided a new and economically attractive route. BASF developed a commercially feasible manufacturing process that was in use by 1897, at which time 19,000 tons of indigo were being produced from plant sources. This had dropped to 1,000 tons by 1914 and continued to contract. By 2011 50,000 tons of synthetic indigo were being produced worldwide.[25]
Dyeing technology
Indigo white (leuco-indigo)
Yarn dyed with indigo dye
Indigo white
Indigo is a challenging dye because it is not soluble in water. To be dissolved, it must undergo a chemical change (reduction). Reduction converts indigo into “white indigo” (leuco-indigo). When a submerged fabric is removed from the dyebath, the white indigo quickly combines with oxygen in the air and reverts to the insoluble, intensely colored indigo. When it first became widely available in Europe in the 16th century, European dyers and printers struggled with indigo because of this distinctive property. It also required several chemical manipulations, some involving toxic materials, and had many opportunities to injure workers. In the 19th century, English poet William Wordsworth referred to the plight of indigo dye workers of his hometown of Cockermouth in his autobiographical poem The Prelude. Speaking of their dire working conditions and the empathy that he felt for them, he wrote:
Doubtless, I should have then made common cause
With some who perished; haply perished too
A poor mistaken and bewildered offering
Unknown to those bare souls of miller blue
A pre-industrial process for production of indigo white, used in Europe, was to dissolve the indigo in stale urine, which contains ammonia. A more convenient reductive agent is zinc. Another pre-industrial method, used in Japan, was to dissolve the indigo in a heated vat in which a culture of thermophilic, anaerobic bacteria was maintained. Some species of such bacteria generate hydrogen as a metabolic product, which convert insoluble indigo into soluble indigo white. Cloth dyed in such a vat was decorated with the techniques of shibori (tie-dye), kasuri, katazome, and tsutsugaki. Examples of clothing and banners dyed with these techniques can be seen in the works of Hokusai and other artists.
Direct printing
Two different methods for the direct application of indigo were developed in England in the 18th century and remained in use well into the 19th century. The first method, known as ‘pencil blue’ because it was most often applied by pencil or brush, could be used to achieve dark hues. Arsenic trisulfide and a thickener were added to the indigo vat. The arsenic compound delayed the oxidation of the indigo long enough to paint the dye onto fabrics.
Pot of freeze-dried indigo dye
The second method was known as ‘China blue’ due to its resemblance to Chinese blue-and-white porcelain. Instead of using an indigo solution directly, the process involved printing the insoluble form of indigo onto the fabric. The indigo was then reduced in a sequence of baths of iron(II) sulfate, with air-oxidation between each immersion. The China blue process could make sharp designs, but it could not produce the dark hues possible with the pencil blue method.
Around 1880, the ‘glucose process’ was developed. It finally enabled the direct printing of indigo onto fabric and could produce inexpensive dark indigo prints unattainable with the China blue method.
Since 2004, freeze-dried indigo, or instant indigo, has become available. In this method, the indigo has already been reduced, and then freeze-dried into a crystal. The crystals are added to warm water to create the dye pot. As in a standard indigo dye pot, care has to be taken to avoid mixing in oxygen. Freeze-dried indigo is simple to use, and the crystals can be stored indefinitely as long as they are not exposed to moisture.[26]
The molecule absorbs light in the orange part of the spectrum (λmax = 613 nm).[27] The compound owes its deep color to the conjugation of the double bonds, i.e. the double bonds within the molecule are adjacent and the molecule is planar. In indigo white, the conjugation is interrupted because the molecule is non-planar.
Indigo derivatives
Structure of Tyrian purple
Structure of indigo carmine.
The benzene rings in indigo can be modified to give a variety of related dyestuffs. Thioindigo, where the two NH groups are replaced by S atoms, is deep red. Tyrian purple is a dull purple dye that is secreted by a common Mediterranean snail. It was highly prized in antiquity. In 1909, its structure was shown to be 6,6′-dibromoindigo (red). 6-bromoindigo (purple) is a component as well.[28] It has never been produced on a commercial basis. The related Ciba blue (5,7,5′,7′-tetrabromoindigo) is, however, of commercial value.
Indigo and its derivatives featuring intra- and intermolecular hydrogen bonding have very low solubility in organic solvents. They can be made soluble using transient protecting groups such as the tBOC group, which suppresses intermolecular bonding.[29] Heating of the tBOC indigo results in efficient thermal deprotection and regeneration of the parent H-bonded pigment.
Treatment with sulfuric acid converts indigo into a blue-green derivative called indigo carmine (sulfonated indigo). It became available in the mid-18th century. It is used as a colorant for food, pharmaceuticals, and cosmetics.
Indigo as an organic semiconductor
Indigo and some of its derivatives are known to be ambipolar organic semiconductors when deposited as thin films by vacuum evaporation.[30]
Safety and the environment
Indigo has a low oral toxicity, with an LD50 of 5000 mg/kg in mammals.[1] In 2009, large spills of blue dyes had been reported downstream of a blue jeans manufacturer in Lesotho.[31]
^Quoted in Hentschel, Klaus (2002). Mapping the spectrum: techniques of visual representation in research and teaching. Oxford, England: Oxford University Press. p. 28. ISBN978-0-19-850953-0.
^Andrea Feeser, Red, White, and Black Make Blue: Indigo in the Fabric of Colonial South Carolina Life (University of Georgia Press; 2013)
^Jones, Claude E. (1958). “Charles Woodmason as a Poet”. The South Carolina Historical Magazine. 59 (4): 189–194.
^David S. Shields. Oracles of Empire: Poetry, Politics, and Commerce in British America, 1690-1750. (Chicago: University of Chicago Press, 2010), pp. 69, 249
^Walter B. Edgar, ed. The South Carolina Encyclopedia. Columbia, SC: University of South Carolina Press, 2006), p. 9.
^Schoenbrun, David (1976). Triumph in Paris: The Exploits of Benjamin Franklin. New York: Harper & Row. p. 51. ISBN978-0-06-013854-7.
^David H. Rembert, Jr. (1979). “The indigo of commerce in colonial North America”. Economic Botany. 33 (2): 128–134. doi:10.1007/BF02858281. S2CID2488865.
^“History of Indigo & Indigo Dyeing”. wildcolours.co.uk. Wild Colours and natural Dyes. Retrieved 30 December 2015. Indigo was often referred to as Blue Gold as it was an ideal trading commodity; high value, compact and long lasting
^Adolf Baeyer (1883) “Ueber die Verbindungen der Indigogruppe” [On the compounds of the indigo group], Berichte der Deutschen chemischen Gesellschaft zu Berlin, 16 : 2188-2204 ; see especially p. 2204.
^Wouten, J.; Verhecken, A. (1991). “High-performance liquid chromatography of blue and purple indigoid natural dyes”. Journal of the Society of Dyers and Colourists. 107: 266–269.
^Ramig, Keith; Lavinda, Olga; Szalda, David J.; Mironova, Irina; Karimi, Sasan; Pozzi, Federica; Shah, Nilam; Samson, Jacopo; Ajiki, Hiroko; Massa, Lou; Mantzouris, Dimitrios; Karapanagiotis, Ioannis; Cooksey, Christopher (June 2015). “The nature of thermochromic effects in dyeings with indigo, 6-bromoindigo, and 6,6′-dibromoindigo, components of Tyrian purple”. Dyes and Pigments. 117: 37–48. doi:10.1016/j.dyepig.2015.01.025.
^Głowacki, Eric Daniel; Voss, Gundula; Demirak, Kadir; Havlicek, Marek; Sünger, Nevsal; et al. (2013). “A facile protection–deprotection route for obtaining indigo pigments as thin films and their applications in organic bulk heterojunctions”. Chemical Communications. 49 (54): 6063–6065. doi:10.1039/C3CC42889C. PMID23723050.
^Irimia-Vladu, Mihai; Głowacki, Eric D.; Troshin, Pavel A.; Schwabegger, Günther; Leonat, Lucia; Susarova, Diana K.; Krystal, Olga; Ullah, Mujeeb; Kanbur, Yasin; Bodea, Marius A.; Razumov, Vladimir F.; Sitter, Helmut; Bauer, Siegfried; Sarıçiftçi, Niyazi Serdar (2012). “Indigo – A Natural Pigment for High Performance Ambipolar Organic Field Effect Transistors and Circuits”. Advanced Materials. 24 (3): 375–80. doi:10.1002/adma.201102619. PMID22109816.
^Denison MS, Nagy SR (2003). “Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals”. Annu. Rev. Pharmacol. Toxicol. 43: 309–34. doi:10.1146/annurev.pharmtox.43.100901.135828. PMID12540743.
It is traditionally regarded as a color in the visible spectrum, as well as one of the seven colors of the rainbow: the color between blue and violet; however, sources differ as to its actual position in the electromagnetic spectrum.
The first known recorded use of indigo as a color name in English was in 1289.[3]
Indigofera tinctoria and related species were cultivated in East Asia, Egypt, India, and Peru in antiquity. The earliest direct evidence for the use of indigo dates to around 4000 BC and comes from Huaca Prieta, in contemporary Peru.[4]Pliny the Elder mentions India as the source of the dye after which it was named.[5] It was imported from there in small quantities via the Silk Road.[6]
Spanish explorers discovered an American species of indigo and began to cultivate the product in Guatemala. The English and French subsequently began to encourage indigo cultivation in their colonies in the West Indies.[8]
In North America, indigo was introduced by Eliza Lucas into colonial South Carolina, where it became the colony’s second-most important cash crop (after rice).[9] Before the Revolutionary War, indigo accounted for more than one-third of the value of exports from the American colonies.[10]
Blue dye can be made from two different types of plants: the indigo plant, which produces the best results, and from the woad plant Isatis tinctoria, also known as pastel.[11] For a long time, woad was the main source of blue dye in Europe. Woad was replaced by true indigo as trade routes opened up, and both plant sources have now been largely replaced by synthetic dyes.
Classification as a spectral color
Indigo is one of the colors on Newton’s color wheel.
The correspondence of this definition with colors of actual indigo dyes, though, is disputed. Optical scientists Hardy and Perrin list indigo as between 445[15] and 464 nm wavelength,[16] which occupies a spectrum segment from roughly the color wheel (RGB) blue extending to the long-wave side, towards azure.
Isaac Newton introduced indigo as one of the seven base colors of his work. In the mid-1660s, when Newton bought a pair of prisms at a fair near Cambridge, the East India Company had begun importing indigo dye into England,[17] supplanting the homegrown woad as source of blue dye. In a pivotal experiment in the history of optics, the young Newton shone a narrow beam of sunlight through a prism to produce a rainbow-like band of colors on the wall. In describing this optical spectrum, Newton acknowledged that the spectrum had a continuum of colors, but named seven: “The originall or primary colours are Red, yellow, Green, Blew, & a violet purple; together with Orang, Indico, & an indefinite varietie of intermediate gradations.”[18] He linked the seven prismatic colors to the seven notes of a western major scale,[19] as shown in his color wheel, with orange and indigo as the semitones. Having decided upon seven colors, he asked a friend to repeatedly divide up the spectrum that was projected from the prism onto the wall:
Newton’s observation of prismatic colors: Comparing this to a color image of the visible light spectrum shows that indigo corresponds to blue, while blue corresponds to cyan.
I desired a friend to draw with a pencil lines cross the image, or pillar of colours, where every one of the seven aforenamed colours was most full and brisk, and also where he judged the truest confines of them to be, whilst I held the paper so, that the said image might fall within a certain compass marked on it. And this I did, partly because my own eyes are not very critical in distinguishing colours, partly because another, to whom I had not communicated my thoughts about this matter, could have nothing but his eyes to determine his fancy in making those marks.[20]
Traditional seven colors of the rainbow
Indigo is therefore counted as one of the traditional colors of the rainbow, the order of which is given by the mnemonics “Richard of York gave battle in vain” and Roy G. Biv. James Clerk Maxwell and Hermann von Helmholtz accepted indigo as an appropriate name for the color flanking violet in the spectrum.[21]
Later scientists concluded that Newton named the colors differently from current usage.[22][23] According to Gary Waldman, “A careful reading of Newton’s work indicates that the color he called indigo, we would normally call blue; his blue is then what we would name blue-green, cyan or light blue.”[24] If this is true, Newton’s seven spectral colors would have been:
The human eye does not readily differentiate hues in the wavelengths between what are now called blue and violet. If this is where Newton meant indigo to lie, most individuals would have difficulty distinguishing indigo from its neighbors. According to Isaac Asimov, “It is customary to list indigo as a color lying between blue and violet, but it has never seemed to me that indigo is worth the dignity of being considered a separate color. To my eyes, it seems merely deep blue.”[25]
Modern color scientists typically divide the spectrum between violet and blue at about 450 nm, with no indigo.[26][27]
Distinction among the four major tones of indigo
Like many other colors (orange, rose, and violet are the best-known), indigo gets its name from an object in the natural world—the plant named indigo once used for dyeing cloth (see also Indigo dye).
The color “electric indigo” is a bright and saturated color between the traditional indigo and violet. This is the brightest color indigo that can be approximated on a computer screen; it is a color located between the (primary) blue and the color violet of the RGB color wheel.
The web color blue violet or deep indigo is a tone of indigo brighter than pigment indigo, but not as bright as electric indigo.
The color pigment indigo is equivalent to the web color indigo and approximates the color indigo that is usually reproduced in pigments and colored pencils.
The color of indigo dye is a different color from either spectrum indigo or pigment indigo. This is the actual color of the dye. A vat full of this dye is a darker color, approximating the web color midnight blue.
Below are displayed these four major tones of indigo.
“Electric indigo” is brighter than the pigment indigo reproduced below. When plotted on the CIE chromaticity diagram, this color is at 435 nanometers, in the middle of the portion of the spectrum traditionally considered indigo, i.e., between 450 and 420 nanometers. This color is only an approximation of spectral indigo, since actual spectral colors are outside the gamut of the sRGB color system.
B: Normalized to [0–255] (byte) H: Normalized to [0–100] (hundred)
At right is displayed the web color “blue-violet”, a color intermediate in brightness between electric indigo and pigment indigo. It is also known as “deep indigo”.
B: Normalized to [0–255] (byte) H: Normalized to [0–100] (hundred)
The color box on the right displays the web color indigo, the color indigo as it would be reproduced by artists’ paints as opposed to the brighter indigo above (electric indigo) that is possible to reproduce on a computer screen. Its hue is closer to violet than to indigo dye for which the color is named. Pigment indigo can be obtained by mixing 55% pigment cyan with about 45% pigment magenta.
Compare the subtractive colors to the additive colors in the two primary color charts in the article on primary colors to see the distinction between electric colors as reproducible from light on a computer screen (additive colors) and the pigment colors reproducible with pigments (subtractive colors); the additive colors are significantly brighter because they are produced from light instead of pigment.
Web color indigo represents the way the color indigo was always reproduced in pigments, paints, or colored pencils in the 1950s. By the 1970s, because of the advent of psychedelic art, artists became accustomed to brighter pigments. Pigments called “bright indigo” or “bright blue-violet” (the pigment equivalent of the electric indigo reproduced in the section above) became available in artists’ pigments and colored pencils.
B: Normalized to [0–255] (byte) H: Normalized to [0–100] (hundred)
‘Tropical Indigo’ is the color that is called añil in the Guía de coloraciones (Guide to colorations) by Rosa Gallego and Juan Carlos Sanz, a color dictionary published in 2005 that is widely popular in the Hispanophone realm.
B: Normalized to [0–255] (byte) H: Normalized to [0–100] (hundred)
Indigo dye is a greenish dark blue color, obtained from either the leaves of the tropical Indigo plant (Indigofera), or from woad (Isatis tinctoria), or the Chinese indigo (Persicaria tinctoria). Many societies make use of the Indigofera plant for producing different shades of blue. Cloth that is repeatedly boiled in an indigo dye bath-solution (boiled and left to dry, boiled and left to dry, etc.), the blue pigment becomes darker on the cloth. After dyeing, the cloth is hung in the open air to dry.
A Native American woman described the process used by the Cherokee Indians when extracting the dye:
We raised our indigo which we cut in the morning while the dew was still on it; then we put it in a tub and soaked it overnight, and the next day we foamed it up by beating it with a gourd. We let it stand overnight again, and the next day rubbed tallow on our hands to kill the foam. Afterwards, we poured the water off, and the sediment left in the bottom we would pour into a pitcher or crock to let it get dry, and then we would put it into a poke made of cloth (i.e. sack made of coarse cloth) and then when we wanted any of it to dye [there]with, we would take the dry indigo.[30][31]
In Sa Pa, Vietnam, the tropical Indigo (Indigo tinctoria) leaves are harvested and, while still fresh, placed inside a tub of room-temperature to lukewarm water where they are left to sit for 3 to 4 days and allowed to ferment, until the water turns green. Afterwards, crushed limestone (pickling lime) is added to the water, at which time the water with the leaves are vigorously agitated for 15 to 20 minutes, until the water turns blue. The blue pigment settles as sediment at the bottom of the tub. The sediment is scooped out and stored. When dyeing cloth, the pigment is then boiled in a vat of water; the cloth (usually made from yarns of hemp) is inserted into the vat for absorbing the dye. After hanging out to dry, the boiling process is repeated as often as needed to produce a darker color.
The eastern indigo snake, Drymarchon couperi, of the southeastern United States, is a dark blue/black.
In culture
Literature
Marina Warner’s novel Indigo (1992) is a retelling of Shakespeare’s The Tempest and features the production of indigo dye by Sycorax.
Computer graphics
Electric indigo is sometimes used as a glow color for computer graphics lighting, possibly because it seems to change color from indigo to lavender when blended with white.
Dyes
Indigo is created in potholes carved in pumice “tufgrond” in Karoland, Sumatra
Indigo dye was used to dye denim, giving the original ‘blue jeans‘ their distinctive colour.
Guatemala, as of 1778, was considered one of the world’s foremost providers of indigo.[34]
In Mexico, indigo is known as añil.[35] After silver, and cochineal to produce red, añil was the most important product exported by historical Mexico.[36]
The use of añil is survived in the Philippines, particularly in the Visayas and Mindanao. The powder dye is mixed with vinegar to be applied to the cheek of a person suffering from mumps.[citation needed]
Food
Scientists discovered in 2008 that when a banana becomes ripe, it glows bright indigo under a black light. Some insects, as well as birds, see into the ultraviolet, because they are tetrachromats and can use this information to tell when a banana is ready to eat. The glow is the result of a chemical created as the green chlorophyll in the peel breaks down.[37]
Military
The French Army adopted dark blue indigo at the time of the French Revolution, as a replacement for the white uniforms previously worn by the Royal infantry regiments. In 1806, Napoleon decided to restore the white coats because of shortages of indigo dye imposed by the British continental blockade. However, the greater practicability of the blue color led to its retention, and indigo remained the dominant color of French military coats until 1914.
The color electric indigo is used in New Age philosophy to symbolically represent the sixth chakra (called Ajna), which is said to include the third eye. This chakra is believed to be related to intuition and gnosis (spiritual knowledge).[39][40]
In Paganism, it represents emotion, fluidity, insight, and expressiveness. It is used to spiritually heal.[43]
Indigo synthesis
Aims of the experiment Synthesis of the organic product indigo Explanation of the chromaticity of indigo. Folloing the Bayer-Drewsen reaction mechanism. Calculation of yield.
Principles Indigo synthesis was discovered in 1870 by Adolph von Bayer. It made it possible, for the first time, to synthetically produce indigo, one of the oldest and most important natural dyes. Today, dyeing of jeans is still the main use of indigo. With an annual worldwide production of 30,000 tons,indigo is still the most used textile dye
The chromaticity of indigo can be explained by the formation of a conjugated π system with a total of 22 π electrons. In addition, there are 18 electrons from 9 double bonds and 4 electrons from free electron pairs at the nitrogen atoms. In organic dyes, it is the P orbitals which run perpendicular to the core bond axis that form the conjugated π systems if they lie in a common plane and are adjacent to one another. The electrons distribute across the bonding molecular orbitals and are delocalised over the entire π system.
Through electromagnetic radiation, electrons can be lifted from the bonding molecular orbitals to antibonding molecular orbitals if the energy of the irradiating light quanta corresponds to the energy of the orbital transition. This process is called absorption. The wavelength at which a substance absorbs the most light is determined by the energy difference between the highest occupied and the lowest unoccupied molecular orbital. The more orbitals involved in a π system, the more the energy states that exist vary and the lower the energetic gap is between the highest occupied and the lowest unoccupied orbital. As the size of the π system increases, the absorption maximum of a dye shifts to the longer-wave spectral range. The absorbed portion of light is removed from the spectrum of emitted light. Indigo absorbs light in the yellow spectral range. The emitted light appears to us in the complementary colour blue. Through structural modification of indigo, other colour shades than blue, which is characteristic of indigo, can be generated. The group of substances derived from indigo is called indigoid dyes. In the experiment presented here, indigo will be produced according to the Bayer-Drewsen reaction from 2-nitrobenzaldehyde. In the evaluation, the reaction mechanism will be elucidated and the yield is calculated. The use of indigo in dyeing is presented in experiment C5.2.4.1.
Risk assessment When carrying out the experiment, wear goggles, an apron and gloves. Be careful in particular when adding the sodium hydroxide pellets, as they are very corrosive. Keep the bottles of organic solvent away from possible flame sources.
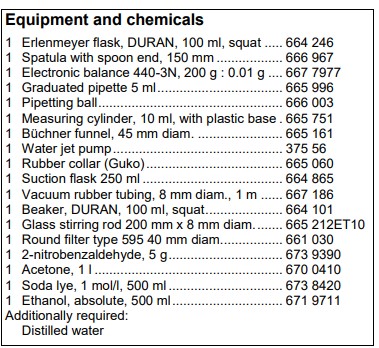
Set-up and preparation of the experiment Synthesis of indigo In a 100 ml Erlenmeyer flask, 1 g of 2-nitrobenzaldehyde is weighed out. Acetone, 1 N sodium hydroxide and distilled water are prepared. Also, a 5 ml graduated pipette with a pipetting ball is provided and a 10 ml measuring cylinder. The porcelain Büchner funnel is inserted in to the suction flask with the rubber collar. The suction flask is then connected to the water jet pump through a tube. A type 595 round filter is placed in the Büchner funnel in such a way that all holes of the funnel are covered. A 100 ml beaker is prepared with 50 ml of ethanol.
Dying with indigo To dye the material, a 150 ml beaker is filled with 100 ml of distilled water and placed on a magnetic stirrer with hotplate. 2 g of sodium dithionite is weighed out onto a watch glass. Sodium hydroxide pellets and ethanol are also needed.
Performing the experiment
Synthesis of indigo The weighed out 2-nitrobenzaldehyde is dissolved in 3 ml of acetone. Then, 3 ml of distilled water and 1 ml of 1N soda lye are added. The solution changes colour to dark brown in the process. After 5 minutes, the solution is filtered. To do so, the water jet pump is first turned on. The filter is made wet with a bit of ethanol. Note: Check to see that the filter is situated correctly! All holes of the funnel must be covered by filter paper. Only then are the contents of the Erlenmeyer flask poured over the filter in small steps. Contents remaining in the Erlenmeyer flask are flushed out with ethanol and also added to the Büchner funnel. After the liquid in the Erlenmeyer flask is filtered, the residue in the Büchner funnel is washed again with a bit of ethanol. Then the pump is turned off. The residue obtained will still look a bit brown, but can be used for dying.
Observation 1. After adding the sodium hydroxide to 2-nitrobenzaldehyde and acetone, the solution turns dark brown. 2. During nutsch filtering, a blue-brown mixture is obtained. 3. The dried indigo weighs about 1.1 g.
Result of the experiment
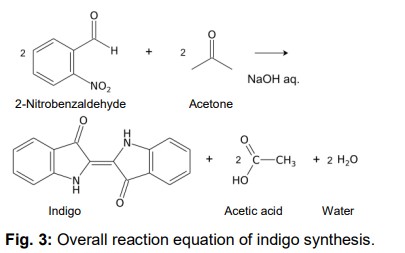
Indigo synthesis mechanism In indigo synthesis 2 molecules of acetone formally react with 2 molecules of 2-nitrobenzaldehyde with splitting of 2 molecules of acetic acid and 2 molecules of water to form indigo. molecules of acetic acid and 2 molecules of water to form indigo.
The first step of the mechanism is an aldol addition. The sodium hydroxide causes an acidic proton to split off. The acetone can then attack the carbonyl group of the 2-nitrobenzaldehyde as a nucleophile. The result of the aldol reaction is an aldol (Fig. 4).
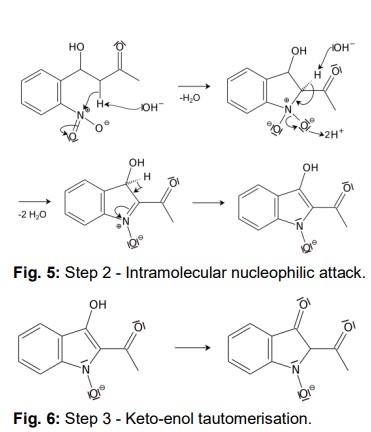
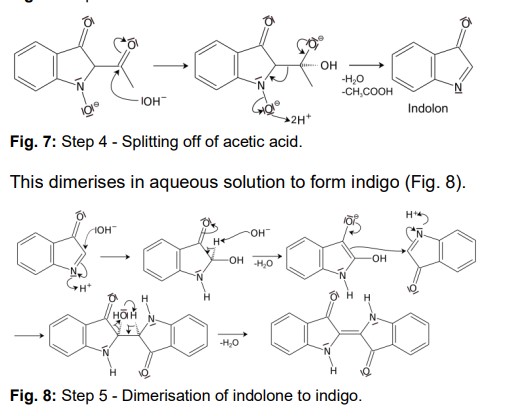
Another acidic proton can be split off. In the second step, the free electron pair at the carbon atom attacks the nitro group nucleophilically in an intramolecular reaction. The splitting of the third acidic proton leads to the formation of a double bond at the nitrogen and enables the splitting of one of the oxygens as water. After the formation of the double bond, a fourth acidic proton can be split off. A double bond is generated next to the hydroxide group. The electron pair of the double bond at the nitrogen travels to the previously formally positively charged nitrogen (Fig. 5). The third step is a tautomeric conversion of the enol to the keto form (Fig. 6). In the fourth step, a hydroxide ion nucleophilically attacks one of the carbonyl groups. Acetic acid and water are split off. The instabile orange indolone is produced as an intermediate synthesis product (Fig. 7).
The brownish colour after addition of sodium hydroxide is explained as a mixed colour of orange indolone and blue indigo since the dimerisation does not initially proceed to completion. Determination of Yield For the calculation of yield the theoretically possible amount of indigo is compared to the actual isolated amount. Limited starting material in this case I 2-Nitrobenzaldehyde. The other substances are present in excess. 2 g Nitrobenzaldehyde (M = 151,12 g/mol) are 13,2 mmol. Looking at the reaction equation (Fig 2), 2 molecules of 2-Nitrobenzaldehyde yield 1 molecule of indigo. The maximal amount is thus 13,2 / 2 mmol = 6,6 mmol Indigo (M = 262,27 g/mol). In the filter, a maximum of 6,6 mmol ∙ 262,27 g/mol = 1,7 g indigo can be present. In the experiment, 1.1 g Indigo were isolated. Thus, the yield is 65%. Cleaning and disposal The wash water contains ethanol, therefore, it must be added to the container for organic solvent waste. The rest of the vat can be added to the container for inorganic solvent waste.
See also
Baptisia (false indigo), a genus of flowering plants
^Robin J. H. Clark, Christopher J. Cooksey, Marcus A. M. Daniels, Robert Withnall: “Indigo, woad, and Tyrian Purple: important vat dyes from antiquity to the present”, Endeavour 17/4 (1993), 191–199.
^Ἰνδικός in Henry George Liddell. Robert Scott. A Greek-English Lexicon. revised and augmented throughout by. Sir Henry Stuart Jones. with the assistance of. Roderick McKenzie. Oxford. Clarendon Press. 1940; English indigo since the 17th century, changed from 16th-century indico.
^Pritchard, James (2004). In Search of Empire: The French in the Americas, 1670–1730. Cambridge: Cambridge University Press. p. 127.
^Ottenheimer, Harriet Joseph (2009). The anthropology of language: an introduction to linguistic anthropology (2nd ed.). Belmont, CA: Wadsworth. p. 29. ISBN978-0-495-50884-7.
^Arthur C. Hardy and Fred H. Perrin. The Principles of Optics.McGraw-Hill Book Co., Inc., New York. 1932.
^Allen, O.N. Allen & Ethel K. (1981). The Leguminosae: a source book of characteristics, uses, and nodulation (null ed.). Madison, Wisc.: University of Wisconsin Press. p. 343. ISBN978-0-299-08400-4.
^Gallego, Rosa; Sanz, Juan Carlos (2005). Guía de coloraciones(Gallego, Rosa; Sanz, Juan Carlos (2005). Guide to Colorations) Madrid: H. Blume. ISBN84-89840-31-8
^Knight, Oliver (1956–57), “History of the Cherokees, 1830–1846”, Chronicles of Oklahoma, Oklahoma City: Oklahoma Historical Society, p. 164, OCLC647927893
^Tansley, David W. Subtle Body: Essence and Shadow 1984 (Art and Cosmos Series–Jill Purce, editor)
^Stevens, Samantha. The Seven Rays: a Universal Guide to the Archangels. City: Insomniac Press, 2004. ISBN1-894663-49-7 pg. 24
^Graham, Lanier F. (editor) The Rainbow Book Berkeley, California:1976 Shambala Publishing and The Fine Arts Museums of San Francisco (Handbook for the Summer 1976 exhibition The Rainbow Art Show which took place primarily at the De Young Museum, but also at other museums) Indigo Pages 152–153 The color indigo is stated to represent intuition.
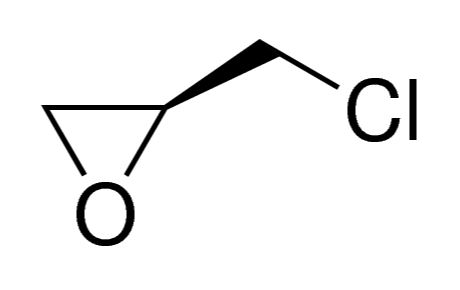
AGGARWWAL EXPORTS (agexpharma.com): S-Epichlorohydrin manufacturer in India
Aggarwwal Exports (www.agexpharma.com) leads the way to become first company in India to manufacture S- Epichlorohydrin (CAS No.: 67843-74-7) in India.
The company holds a cGMP and ISO 9001 registration for the product with an audit ready plant. Aggarwwal Exports – Specialty chemical division’s core strength is Chiral Molecules and S-Epichlorohydrin has various applications such as Rivaroxaban, Linezolid and Brivaracetam. These drugs are being developed quite rapidly all over India.
As China’s reliablility due to Trade War, COVID and pollution problems have arisen; more companies are looking to develop Indian Manufacturers to ensure longevity and robustness of Raw Material Supply.
The company has developed a process from the base level of Racemic Epichlorohydrin and is utilizing a precious metal catalyst to create chirally pure S-Epichlorohydrin and R-Glycidol, which is further processed to make R-Glycidyl Butyrate and S-Glycidyl
Phathalimide
Benefits of purchasing S-Epichlorohydrin from Aggarwwal Exports-
1) 99.5% GC purity and 99.9% Chiral Purity
2) Importing from China and Japan is a tedious process as import duty needs to be
made, lead time is around 2 months as S-Epichlorohydrin is under Hazardous cargo
which is expensive to ship.
3) Pricing is slightly lower than imports but quality is improved.
4) Lead time is 1 week from order confirmation.
5) Most importantly, Make in India.
For any inquiry please contact Aggarwwal Exports:
Email ID: Pranjal@agexpharma.com
Cell no.: +91-9837035981
Pranjal Varshney,V.P. Operations
Aggarwwal Exports
Aggarwwal Exports, B-8 Roshan Bagh, Rampur 244901 (UP) India