












Olopatadine hydrochloride
Cis form, Z Isomer
( Z ) – 1 1 – [ 3 – ( D i m e t h y l a m i n o ) p r opy l i d e n e ] – 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride
ALO 4943A; Allelock; KW 4679; Opatanol; Patanol;
| (11Z)-11-[3-(Dimethylamino)propylidene]-6,11-dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride; | |
| CAS Number: | 140462-76-6 |
| unii 2XG66W44KF | |
| Molecular form.: | C₂₁H₂₄ClNO₃ |
| Appearance: | White Solid |
| Melting Point: | >240˚C (dec.) |
| Mol. Weight: | 373.87 |
Olopatadine hydrochloride is an antihistamine (as well as anticholinergic and mast cell stabilizer), sold as a prescription eye dropmanufactured by Alcon in one of three strengths: 0.7% solution or Pazeo in the US, 0.2% solution or Pataday (also called Patanol Sin some countries), and 0.1% or Patanol (also called Opatanol in some countries). It is used to treat itching associated with allergicconjunctivitis (eye allergies). A decongestant nasal spray formulation is sold as Patanase, which was approved by the FDA on April 15, 2008.[1] It is also available as an oral tablet in Japan under the tradename Allelock, manufactured by Kyowa Hakko Kogyo.[2]
It should not be used to treat irritation caused by contact lenses. The usual dose for Patanol is 1 drop in each affected eye 2 times per day, with 6 to 8 hours between doses. Both Pazeo and Pataday are dosed 1 drop in each eye daily.
There is potential for Olopatadine as a treatment modality for steroid rebound (red skin syndrome).[3]
Olopatadine was developed by Kyowa Hakko Kogyo.[4]
Side Effects
Some known side effects include headache (7% of occurrence), eye burning and/or stinging (5%), blurred vision, dry eyes, foreign body sensation, hyperemia, keratitis, eyelid edema, pruritus, asthenia, sore throat (pharyngitis), rhinitis, sinusitis, and taste perversion.
Synthesis

Olopatadine synthesis:[5]
Patent




PATENT






















PATENT



Journal of the Brazilian Chemical Society
J. Braz. Chem. Soc. vol.25 no.12 São Paulo Dec. 2014
http://dx.doi.org/10.5935/0103-5053.20140255
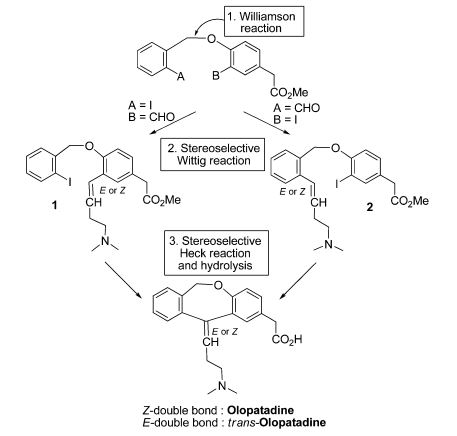
An intramolecular Heck-based cyclization was used as a key step for commendable synthesis of the antihistaminic drug olopatadine (133) and its trans isomer (134).67 Besides the Heck reaction, another vital step in this route was a stereoselective Wittig olefination using a non-stabilized phosphorus ylide that afforded the olefins 135 and 136 (E:Z ratio = 9:1 for 135, for instance). Concerning the Heck reaction, Pd(OAc)2, K2CO3, and NBu4Cl (TBAC) were allowed to react with 135 and 136 at 60 ºC during 24 h, providing the cyclic adducts 137 and 138 with reasonable 60% and 55% yields, respectively. However, it is important to note that in catalytic terms, the results were not encouraging, considering that 20 mol% of palladium was used and a disappointing turnover number (TON) of 3 was observed (Scheme 37).

In relation to the stereochemistry of the Heck products, the above results were not surprising since they were consistent with a syn-insertion of the arylpalladium intermediate (provided by oxidative addition step) at the olefinic moiety followed by a syn β-elimination that afforded the product with the ascribed stereochemistry. Finally, with the cyclic products in hands, the syntheses were completed by alkaline hydrolysis of methyl esters that afforded the target olapatadine and trans-olapatadine.
67 Bosch, J.; Bachs, J.; Gómez, A. M.; Griera, R.; Écija, M.; Amat, M.; J. Org. Chem.2012, 77 , 6340.
SPECTROSCOPY FROM NET
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT,
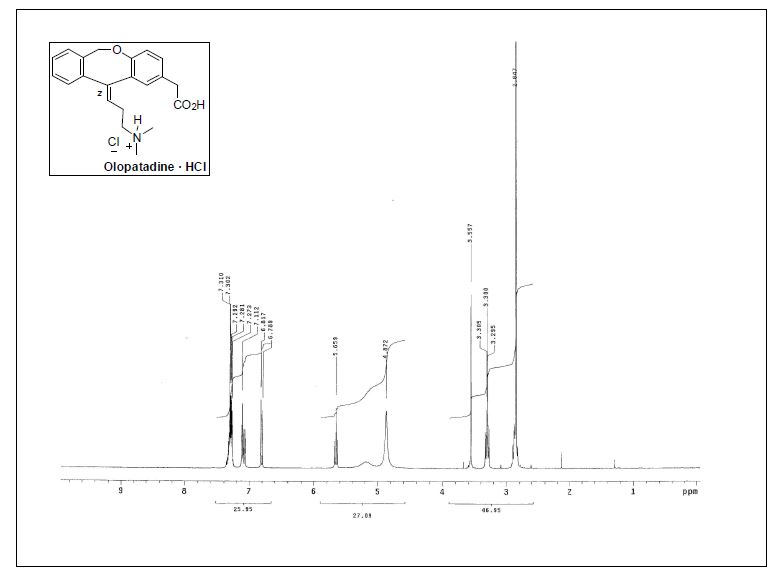
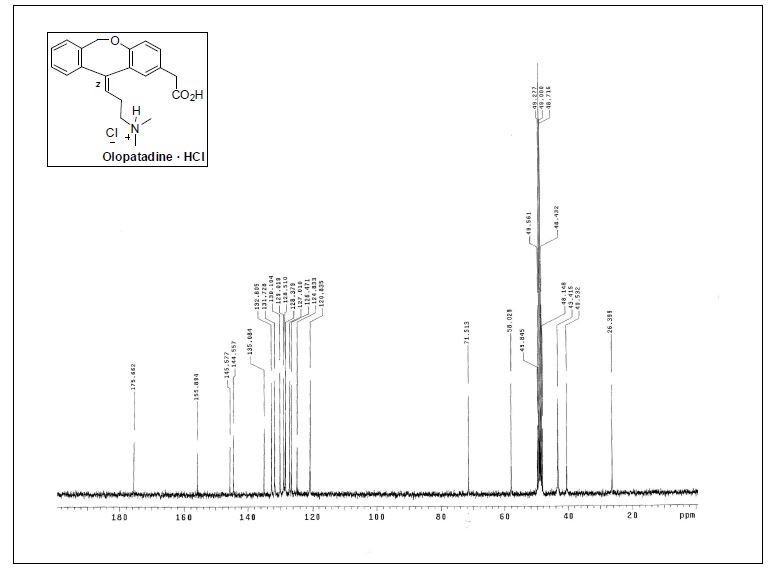
( Z ) – 1 1 – [ 3 – ( D i m e t h y l a m i n o ) p r opy l i d e n e ] – 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride.
Cis-olopatadinehydrochloride /olopatadinehydrochloride
mp 231−233 °C (dec);
1H NMR (300 MHz, CD3OD) δ 2.86 (s, 6H), 2.83−2.91 (m, 2H), 3.28−3.34 (m, 2H), 3.57 (s, 2H), 5.19 (br, 2H), 5.67 (t,J = 7.3 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 7.07−7.13 (m, 2H), 7.26−7.37 (m, 4H);
13C NMR (75.4 MHz, CD3OD) δ 26.4 (CH2), 40.5(CH2), 43.4 (2CH3), 58.0 (CH2), 71.5 (CH2), 120.3 (CH), 124.8 (C),126.5 (CH), 127.0 (CH), 128.4 (C), 128.5 (CH), 129.0 (CH), 130.1(CH), 131.7 (CH), 132.8 (CH), 135.1 (C), 144.5 (C), 145.6 (C),155.9 (C), 175.7 (C);
IR (KBr) 1225, 1491, 1716, 2927 cm−1.
Anal.Calcd for C21H24NClO3·H
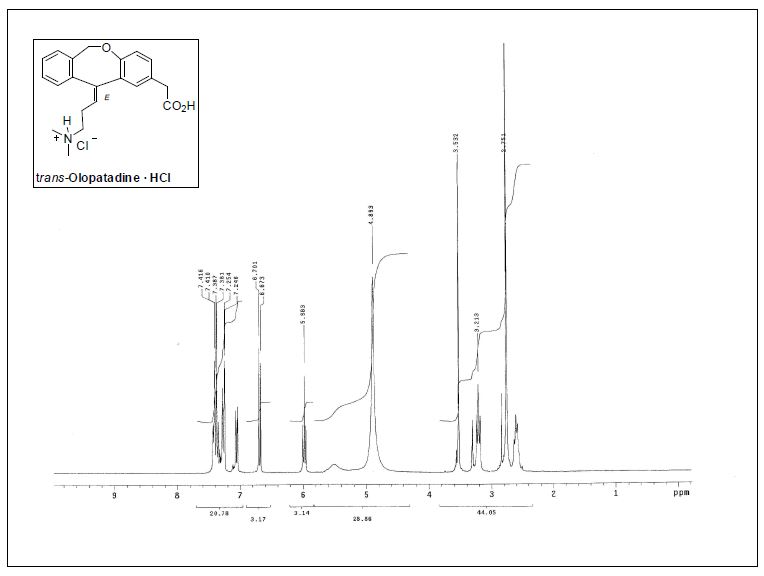
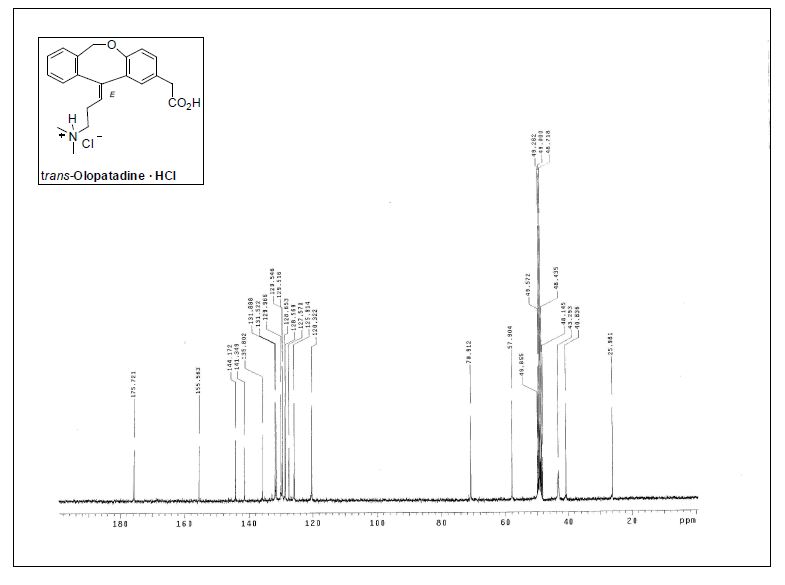
( E ) – 1 1 – [ 3 – ( D i m e thy l ami n o ) p r o p y l i d e n e ] – 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride.
trans-olopatadinehydrochloride
mp 170−173 °C;
1H NMR (300 MHz, CD3OD) δ 2.56−2.63 (m, 2H), 2.75 (s,6H), 3.13 (t, J = 7.6 Hz, 2H), 3.53 (s, 2H), 4.78 (br, 1H), 5.51 (br,
1H), 5.98 (t, J = 7.2 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 7.06 (dd, J =8.3, 2.3 Hz, 1H), 7.25−7.44 (m, 5H);
13C NMR (75.4 MHz, CD3OD)δ 26.0 (CH2), 40.8 (CH2), 43.3 (2CH3), 57.9 (CH2), 70.9 (CH2),120.3 (CH), 125.9 (CH), 127.6 (C), 128.5 (C), 128.6 (CH), 129.5(2CH), 130.0 (CH), 131.5 (CH), 132.0 (CH), 135.8 (C), 141.3 (C),144.2 (C), 155.6 (C), 175.7 (C);
IR (KBr) 1223, 1484, 1725, 2960cm−1.
Anal. Calcd for C21H24NClO3·H2O: C, 64.36; H, 6.69; N, 3.57.
Found: C, 64.66; H, 6.47; N, 3.56.
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT,
References
- Drugs.com, Alcon’s Patanase Nasal Spray Approved by FDA for Treatment of Nasal Allergy Symptoms
- Kyowa Hakko Kogyo Co., Ltd. (2007). “ALLELOCK Tablets 2.5 & ALLELOCK Tablets 5 (English)” (PDF). Retrieved2008-08-10.
- Tamura T; Matsubara M; Hasegawa K; Ohmori K; Karasawa A. (2005). “Olopatadine hydrochloride suppresses the rebound phenomenon after discontinuation of treatment with a topical steroid in mice with chronic contact hypersensitivity.”.
- Kyowa Hakko Kogyo Co., Ltd. (2002). “Company History”.Company Information. Kyowa Hakko Kogyo Co., Ltd. Retrieved16 September 2010.
- Ueno, K.; Kubo, S.; Tagawa, H.; Yoshioka, T.; Tsukada, W.; Tsubokawa, M.; Kojima, H.; Kasahara, A. (1976). “6,11-Dihydro-11-oxodibenz[b,e]oxepinacetic acids with potent antiinflammatory activity”. Journal of Medicinal Chemistry. 19 (7): 941.doi:10.1021/jm00229a017.
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
{(11Z)-11-[3-(dimethylamino)propylidene]-6,11-
dihydrodibenzo[b,e]oxepin-2-yl}acetic acid |
|
| Clinical data | |
| Trade names | Patanol and others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602025 |
| Pregnancy category |
|
| Routes of administration |
Ophthalmic, intranasal, oral |
| Pharmacokinetic data | |
| Biological half-life | 3 hours |
| Identifiers | |
| CAS Number | 113806-05-6 |
| ATC code | S01GX09 (WHO)R01AC08 (WHO) |
| PubChem | CID 5281071 |
| DrugBank | DB00768 |
| ChemSpider | 4444528 |
| UNII | D27V6190PM |
| KEGG | D08293 |
| ChEMBL | CHEMBL1189432 |
| Chemical data | |
| Formula | C21H23NO3 |
| Molar mass | 337.412 g/mol |
/////////////
CN(C)CCC=C1C2=CC=CC=C2COC3=C1C=C(C=C3)CC(=O)O.Cl
Journal of the Brazilian Chemical Society
J. Braz. Chem. Soc. vol.25 no.12 São Paulo Dec. 2014
http://dx.doi.org/10.5935/0103-5053.20140255
The construction of a new bond between sp2– and sp-hybridized carbons is known as the Sonogashira reaction,48and it is nowadays a widely employed methodology for the construction of arylacetylenes.3,49,50 For example, a Sonogashira coupling was employed by the research and development group of Kyowa Hakko Kirin in a new and concise synthetic route for olopatadine hydrochloride (92), a commercial anti-allergic drug that was previously developed by the same company.51
The reported synthesis goes through the Sonogashira reaction between the easy accessible aryl halide 93 and alkyne 94 leading to adduct 95 in 94% yield. This adduct is then subjected to a second metal-catalyzed transformation, a stereospecific palladium-catalyzed intramolecular cyclization, whose optimum conditions were identified based on an elegant and comprehensive Design of Experiments (DoE) investigation to provide 96(Scheme 28).

Elaboration of the cyclization product 96 through aminomethylation and ester hydrolysis followed by acid work-up completes the synthesis of the final target. Although the presented synthetic route is very promising and concise, providing olopatadine hydrochloride in 54% overall yield for 6 steps from commercially available materials, it has so far been reported only on a laboratory scale (5 g for the Sonogashira coupling and 200 mg for the cyclization step).
51 Nishimura, K.; Kinugawa, M.; Org. Process Res. Dev.2012, 16 , 225

Sorry, the comment form is closed at this time.