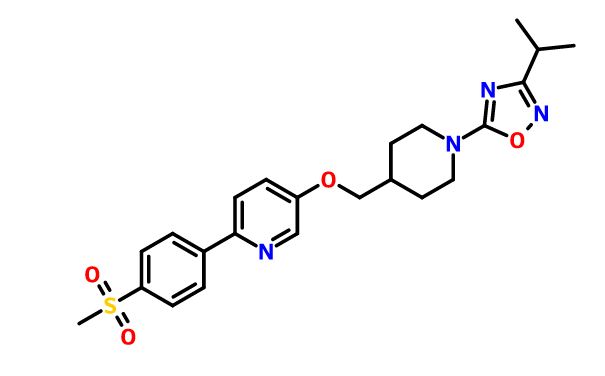
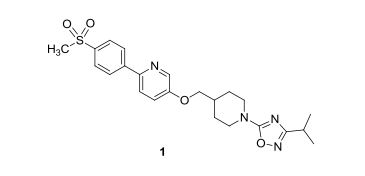
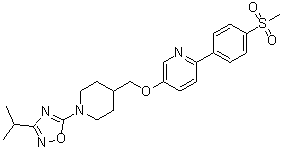
GSK-1292263
CAS 1032823-75-8
3-isopropyl-5-(4-(((6-(4-(methylsulfonyl)phenyl)pyridin-3-yl)oxy)methyl)piperidin-1-yl)-1,2,4-oxadiazole
5-[1-(3-Isopropyl-1,2,4-oxadiazol-5-yl)piperidin-4-ylmethoxy]-2-[4-(methylsulfonyl)phenyl]pyridine
5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine
MF C23H28N4O4S
MW: 456.18313
1292263
GSK-1292263
GSK-1292263A
GSK-263A
Smithkine Beecham Corp, INNOVATOR
GSK-1292263 is a novel GPR119 receptor agonist that is currently under development for the treatment of type 2 diabetes. Treatment of male Sprague-Dawley rats with a single dose of GSK-1292263 (3-30 mg/kg) in the absence of nutrients correlated with increased levels of circulating gastrointestinal peptides; glucagon-like peptide 1 (GLP-1), gastric inhibitory polypeptide (GIP), peptide YY (PYY) and glucagon.
GSK-1292263 had been evaluated in phase II clinical studies at GlaxoSmithKline for the oral treatment of type 2 diabetes and as monotherapy or in combination with sitagliptin for the treatment of dyslipidemia; however no recent development has been reported for this research.
Following administration of glucose in the oral glucose tolerance test (OGTT), greater increases in total GLP-1, GIP and PYY were seen in GSK-1292263-treated rats than in control animals. Despite significant decreases in the glucose AUC, no statistically significant differences in insulin responses and insulin AUC were observed between rats administered GSK-1292263 and those receiving vehicle control.
In the intravenous glucose tolerance test, significant increases in the peak insulin response and insulin AUC(0-15 min) of 30-60% were reported in the GSK-1292263 treatment group, compared with values in the vehicle control cohort. This insulin upregulation correlated with a significant increase in the glucose disposal rate (Brown, K.K. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 407).
The safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of GSK-1292263 were evaluated in a recently completed randomized, placebo-controlled clinical trial in healthy volunteers (ClinicalTrials.gov Identifier NCT00783549).
A total of 69 subjects received single escalating doses of GSK-1292263 (10-400 mg) prior to administration of a 250-mg dose given once daily for 2 and 5 days, which was also evaluated in combination with sitagliptin (100 mg). Treatment with GSK-1292263 at all doses was described as well tolerated, with the most common drug-related effects being mild headache, dizziness, hyperhidrosis, flushing and post-OGTT hypoglycemia.
NMR
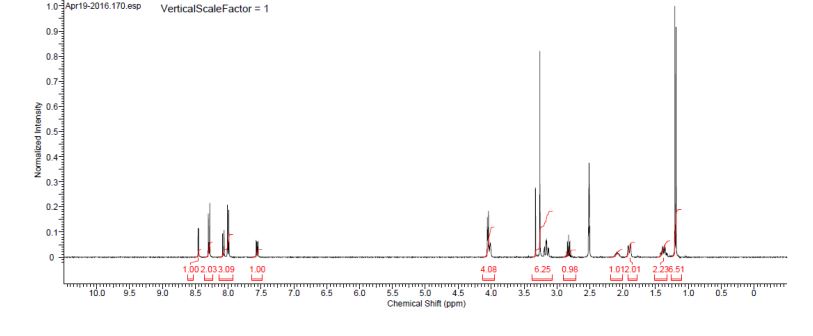
1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 3.0 Hz, 1H), 8.28 (d, J = 8.8 Hz, 2H), 8.06 (d, J = 8.8 Hz, 1H), 7.99 (bd, J = 8.5 Hz, 2H), 7.54 (dd, J = 8.8, 3.0 Hz, 1H), 4.03 (d, J = 6.3 Hz, 2H), 4.03–3.97 (m, 2H), 3.25 (s, 3H), 3.20–3.09 (m, 2H), 2.81 (q, J = 6.7 Hz, 1H), 2.13–2.00 (m, 1H), 1.88 (bd, J = 12.8 H, 2H), 1.42–1.29 (m, 2H), 1.18 (d, J = 7.0 Hz, 6H).
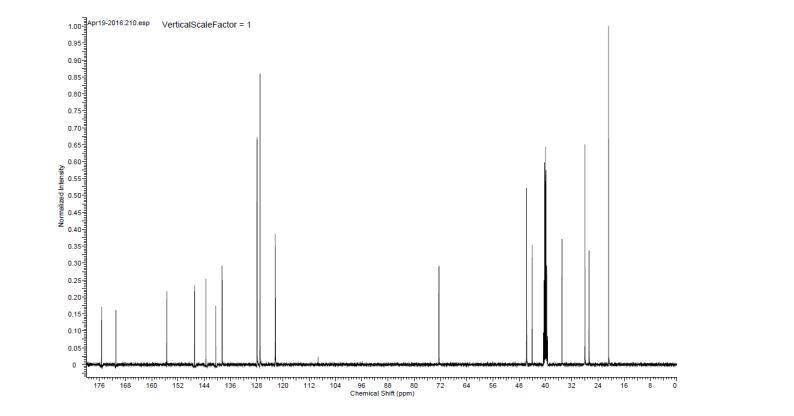
13C NMR (100.6 MHz, DMSO-d6) 175.3, 170.9, 155.5, 147.0, 143.5, 140.5, 138.6, 127.9, 127.0, 122.4, 122.3, 72.5, 45.7, 44.1, 35.0, 28.0, 26.7, 20.8.
HRMS calcd for C23H29N4O4S (M + H)+ 457.1904, found, 457.1900.
Anal. Calcd for C23H28N4O4S: C, 60.51; H, 6.18; N, 12.27. Found: C, 60.64; H, 6.16; N, 12.24.

Hypoglycemia was not reported with the 5-day dosing schedule. Pharmacokinetic profiling revealed dose-proportional AUC and Cmax at single lower doses, but not at single higher ones. Following repeated once-daily dosing (5 days), drug accumulation was observed consistent with a mean half-life of 12-18 hours. A dose-dependent increase in glucose AUC(0-3 h) during OGTT was seen in GSK-1292263-treated subjects. The treatment was also associated with an increase in PYY during the prandial periods.
Coadministration with sitagliptin led to increases in the plasma concentrations of active GLP-1 but reduced the levels of total GLP-1, GIP and PYY. Sitagliptin affected the exposure to GSK-1292263 (50% increase) but GSK-1292263 did not affect sitagliptin exposure. The data support further evaluation of GSK-1292263 for the treatment of type 2 diabetes (Source: Nunez, D.J. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 80-OR).
WO 2008070692
http://www.google.com.au/patents/WO2008070692A2?cl=en
Example 169: 5-[({1 -[3-(1 -Methylethyl)-1,2,4-oxadiazol-5-yl]-4- piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine hydrochloride

Step 1 : A mixture of 6-bromo-3-pyridinol (7 g, 40 mmol), [4-(methylsulfonyl)phenyl]boronic acid (8 g, 40 mmol), 2M Na2CO3 (30 ml_), PdCI2(PPh3)2 (1 g) and DME (60 ml.) under N2 was heated at 80 0C overnight. The reaction was allowed to cool to room temperature and was diluted with EtOAc and water. The resulting precipitate was filtered off and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over MgSO4, filtered and concentrated. The aqueous phase was also concentrated. Each of the residues was recrystallized from MeOH. The solid material from the organic phase recrystallization and the mother liquors from both aqueous and organic recrystallizations were combined, concentrated and purified by chromatography on a silica gel column using 0 to 10% MeOH/CH2CI2 to give 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (2.9 g, 29%) as a tan solid. Step 2: Diisopropyl azodicarboxylate (0.175 ml_, 0.89 mmol) was added dropwise to a solution of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (150 mg, 0.59 mmol), {1-[3-(1- methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 200 mg, 0.89 mmol), PPh3 (233 mg, 0.89 mmol), and THF (10 ml.) at ambient temperature. The mixture was stirred at ambient temperature for 4 h. The mixture was concentrated, and the resulting crude was purified by reverse-phase preparative HPLC using a CH3CN:H2O gradient (10:90 to 100:0) with 0.05% TFA as a modifier, then taken up in CH2CI2 and free-based with saturated NaHCO3 (aq) to give 5-[({1-[3-(1-methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4- (methylsulfonyl)phenyl]pyridine (220 mg) as a white solid. Step 3: A mixture of the resulting white solid (50 mg, 0.1 1 mmol) in THF (3 ml.) was stirred at ambient temperature as 4Λ/ HCI in dioxane (28 μl_) was added dropwise. The resulting white precipitate was filtered, air-dried, then triturated with diethyl ether to give 35 mg (65%) of the title compound as a white solid. 1H NMR (400 MHz, CDCI3): δ 8.46 (d, 1 H, J = 0.7 Hz), 8.18 (bs, 2H), 8.05 (bs, 2H), 7.83 (bs, 1 H), 7.61- 7.45 (m, 1 H), 4.24 (d, 2H, J = 10.4 Hz), 4.00 (d, 2H, J = 0.6 Hz), 3.21-3.03 (m, 5H), 2.89 (m, 1 H), 2.15 (d, 1 H, J = 1.1 Hz), 1.96 (bs, 2H), 1.50 (bs, 2H), 1.28 (d, 6H, J = 6.9 Hz); LRMS (ESI), m/z 457 (M+H).
PATENT
http://www.google.co.ug/patents/US20120077812
Example 100
5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine[0480]
Step 1: A mixture of 2-methylpropanenitrile (100 g, 1.45 mol), hydroxylamine hydrochloride (111 g, 1.59 mol) and NaOH (64 g, 1.59 mol) in EtOH (2 L) and water (500 mL) was stirred at reflux overnight. The mixture was evaporated to dryness and extracted with dichloromethane. The organic extract was dried over Na2SO4 and concentrated to afford the desired N-hydroxy-2-methylpropanimidamide (50 g, 34%).
Step 2: A solution of 4-piperidinemethanol (140 g, 1.22 mol) in CH2Cl2 (1 L) was treated with a slurry of NaHCO3(205 g, 2.44 mol) in water (1.4 L) at 0° C. The mixture was stirred at 0° C. for 15 min, and then charged with a solution of cyanogen bromide in CH2Cl2, (1.34 mol) at 0° C. The reaction mixture was stirred and allowed to warm to ambient temperature, and stirred overnight. The aqueous layer was separated and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4, filtered, and the filtrate was concentrated. The crude product was combined with other batches made similarly and purified by chromatography on a silica gel column to give 300 g of 4-(hydroxymethyl)-1-piperidinecarbonitrile. Step 3: A solution of 1N ZnCl2 in Et2O (182 mL, 182 mmol) was added to a solution of 4-(hydroxymethyl)-1-piperidinecarbonitrile (21.3 g, 152 mmol) and N-hydroxy-2-methylpropanimidamide (18.6 g, 182 mmol) in EtOAc (50 mL) at ambient temperature. The reaction mixture was left at ambient temperature for 30 min, decanted, and was treated with concentrated HCl (45 mL) and ethanol 20 mL). The mixture was heated at reflux for 2 h. The mixture was evaporated to dryness, and the resulting residue was charged with water and the pH was adjusted to basic with K2CO3. The mixture was extracted with EtOAc and the material obtained was combined with 9 other batches prepared similarly and purified by silica gel chromatography to give 150 g of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol.
Step 4: A solution of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Step 3, 174 g, 0.77 mol) and triethylamine (140 mL, 1.0 mol) in dichloromethane (1 L) at 5° C. was treated with a solution of methanesulfonyl chloride (69 mL, 0.89 mol) in dichloromethane (150 mL) over a 1 h period. The mixture was stirred at 5° C. for 30 min, and then was quenched by the addition of water (400 mL). The mixture was stirred for 30 min, and then the organic extract was washed with water (2×400 mL), dried (MgSO4) and concentrated. The residue was treated with heptane (1 L), stirred for 3 h, and the resulting solid was collected by filtration (heptane wash) and air-dried to afford {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (219.7 g, 94%) as an off-white solid. 1NMR (400 MHz, CDCl3): δ 4.21-4.15 (m, 2H), 4.08 (d, 2H, J=6.6 Hz), 3.04 (m, 2H), 3.01 (s, 3H), 2.86 (septet, 1H, J=6.9 Hz), 2.05-1.93 (m, 1H), 1.88-1.81 (m, 2H), 1.43-1.31 (m, 2H), 1.26 (d, 6H, J=6.8 Hz); LRMS (ESI), m/z 304 (M+H).
Step 5: A mixture of 6-bromo-3-pyridinol (36 g, 207 mmol), [4-(methylsulfonyl)phenyl]boronic acid (50 g, 250 mmol), 2M Na2CO3 (315 mL) and DME (500 mL) was degassed with N2 for 30 min, and then Pd(PPh3)4 (12 g, 10 mmol) was added and the mixture was heated at 80° C. for 18 h. The reaction was allowed to cool to room temperature and was diluted with dichloromethane (500 mL) and water (500 mL) and stirred for 30 min. The reaction was filtered and the solids were rinsed with dichloromethane and the aqueous layer was extracted with dichloromethane. The combined organic extracts were extracted with 1N NaOH (2×600 mL), and then cooled to 5° C. and the pH was adjusted to ˜8 with 6N HCl. The resulting precipitate was collected by filtration (water wash) and air-dried to afford a yellow solid. This procedure was repeated and the solids were combined to provide (71.2 g, 68%) of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol. 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H), 8.25 (d, 1H, J=2.7 Hz), 8.21 (d, 2H, J=8.5 Hz), 8.00-7.90 (m, 3H), 7.27 (dd, 1H, Ja=8.7 Hz, Jb=2.8 Hz), 3.21 (s, 3H); LRMS (ESI), m/z 250 (M+H).
Step 6: A mixture of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (82.3 g, 271 mmol), 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (71.0 g, 285 mmol), powdered potassium carbonate (118 g, 855 mmol) and N,N-dimethylformamide (750 mL) was mechanically stirred and heated at 80° C. under nitrogen for 20 h. The reaction was cooled to ambient temperature, poured onto ice water (3 L) and allowed to stand for 1 h. The resulting solid was filtered, rinsed with water (2×500 mL) and air-dried. The solid was taken up in dichloromethane (300 mL) and methanol (500 mL). The dichloromethane was slowly removed via rotovap at 55° C. The methanol solution was allowed to stand at ambient temperature for 16 h. The resulting crystalline solid was filtered, rinsed with cold methanol and dried under vacuum at 60° C. for 18 h to afford the desired product (105.7 g, 84%) as a light tan solid. 1H NMR (400 MHz, CDCl3): δ 8.41 (d, 1H, J=2.8 Hz), 8.13 (d, 2H, J=8.6 Hz), 8.01 (d, 2H, J=8.6 Hz), 7.74 (d, 1H, J=8.7 Hz), 7.29 (dd, 1H, Ja=8.7 Hz, Jb=3.0 Hz), 4.24 (d, 2H, J=13.1 Hz), 3.95 (d, 2H, J=6.2 Hz), 3.17-3.04 (m, 5H), 2.94-2.84 (m, 1H), 2.11 (bs, 1H), 1.97 (d, 2H, J=12.6 Hz), 1.54-1.42 (m, 2H), 1.29 (d, 6H, J=7.0 Hz); LRMS (ESI), m/z 457 (M+H).
Alternative preparation: Step 1: 2-Bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 29%) was prepared as a white solid from {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 348 mg, 2.0 mmol), 6-bromo-3-pyridinol (348 mg, 2.0 mmol) and Ph3P (629 mg, 2.4 mmol) in THF (5 mL) followed by diisopropyl azodicarboxylate (0.51 mL, 2.6 mmol) in a manner similar to Example 1, Step 2. 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.37 (d, 1H, J=8.8 Hz), 7.08 (d, 1H, J=8.8 Hz), 4.26-4.16 (m, 2H), 3.85 (d, 2H, J=6.2 Hz), 3.14-3.04 (m, 2H), 2.95-2.76 (m, 1H), 2.11-1.96 (m, 1H), 1.98-1.88 (m, 2H), 1.52-1.36 (m, 2H), 1.28 (d, 6H, J=6.9 Hz); LRMS (ESI), m/z 381/383 (M+H).
Step 2: 5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine (51 mg, 21%) was prepared from 2-bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 0.52 mmol), [4-(methylsulfonyl)phenyl]boronic acid (105 mg, 0.52 mmol), 2M Na2CO3 (5 mL), Pd(PPh3)4 (50 mg, 0.04 mmol) and DME (5 mL) in a manner similar to Example 21, Step 3.
Paper
Development of Large-Scale Routes to Potent GPR119 Receptor Agonists
Richard T. Matsuoka*†, Eric E. Boros#, Andrew D. Brown†, Kae M. Bullock†, Will L. Canoy‡, Andrew J. Carpenter#, Jeremy D. Cobb†, Shannon E. Condon†, Nicole M. Deschamps†, Vassil I. Elitzin†, Greg Erickson†,Jing M. Fang#, David H. Igo§, Biren K. Joshi‡, Istvan W. Kaldor#, Mark B. Mitchell†, Gregory E. Peckham#, Daniel W. Reynolds‡, Matthew C. Salmon†, Matthew J. Sharp†, Elie A. Tabet#, Jennifer F. Toczko†, Lianming Michael Wu‡, and Xiao-ming M. Zhou†
†API Chemistry Department, ‡Analytical Science & Development Department, #Medicinal Chemistry Department, and§Particle Sciences and Engineering Department, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
Org. Process Res. Dev., Article ASAP
Publication Date (Web): July 13, 2016
Copyright © 2016 American Chemical Society
Abstract
Practical and scalable syntheses were developed that were used to prepare multikilogram batches of GSK1292263A (1) and GSK2041706A (15), two potent G protein-coupled receptor 119 (GPR119) agonists. Both syntheses employed relatively cheap and readily available starting materials, and both took advantage of an SNAr synthetic strategy.
///////////1292263, GSK-1292263, GSK-1292263A, GSK-263A, GSK1292263, GSK1292263A, GSK 1292263, GSK 1292263A, GSK 263A, GSK263A, 1032823-75-8
O=S(C1=CC=C(C2=CC=C(OCC3CCN(C4=NC(C(C)C)=NO4)CC3)C=N2)C=C1)(C)=O














Sorry, the comment form is closed at this time.