











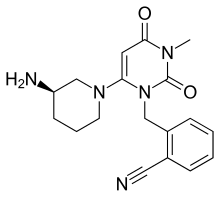
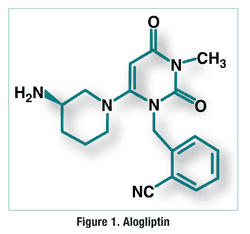
ALOGLIPTIN
Alogliptin is a potent, selective inhibitor of DPP-4 with IC50 of <10 nM, exhibits greater than 10,000-fold selectivity over DPP-8 and DPP-9.
Alogliptin (trade name Nesina in the US[1] and Vipidia in Europe[2]) is an orally administered anti-diabetic drug in the DPP-4 inhibitor class,[3] developed by Syrrx, a company which was acquired by Takeda Pharmaceutical Company in 2005. Like other medications for the treatment of Type 2 diabetes, alogliptin does not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of causing hypoglycemia, and exhibits relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in patients whose diabetes cannot adequately be controlled with metformin alone.[4]
Clinical study
Alogliptin is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide). [5]
A randomized clinical trial reporting in 2011 aimed to determine the efficacy and safety of alogliptin versus placebo and vogliboseamong newly diagnosed Type 2 diabetes patients in Japan. The main outcome indicated that alogliptin was statistically superior to both comparitors.[6]
A randomized clinical trial reporting in 2012 aimed to demonstrate that alogliptin was “non-inferior” to a “very low fat/calorie traditional Japanese diet” among newly diagnosed Type 2 diabetes patients in Japan. The outcome indicated that both the drug and dietary treatments comparably impacted indicators of the diabetic condition, such as HbA1c levels and glycemic efficacy. The drug treatment had its impact without changing body mass index (BMI), but the dietary treatment was accompanied by a significant reduction in the BMI.[7]
A randomized clinical trial reporting in 2011 aimed to demonstrate the efficacy of alogliptin as an add-on agent in combination withmetformin and pioglitazone versus simply increasing the dosage of pioglitazone in combination with metformin; in other words, this was a study to look at a three-agent therapy versus a two-agent therapy. The outcome of this study suggested that the addition of alogliptin to metformin and pioglitazone provided superior impact on diabetes biomarkers (e.g. HbA1c) than increasing the dose of pioglitazone in a two agent therapy with metformin.[8]
Reported adverse events
Adverse events appear to be restricted to mild hypoglycemia based on clinical studies.[6][7][8]
Alogliptin is not associated with increased weight, increased risk of cardiovasular events, or heart failure.[9][10]
Market access
In December 2007, Takeda submitted a New Drug Application (NDA) for alogliptin to the United States Food and Drug Adminiistration (USFDA),[11] after positive results from Phase III clinical trials.[1] In September of 2008, the company also filed for approval in Japan,[12] winning approval in April 2010.[11] The company also filed a Marketing Authorization Application (MAA) elsewhere outside the United States, which was withdrawn in June 2009 needing more data.[12] The first USFDA NDA failed to gain approval and was followed by a pair of NDAs (one for alogliptin and a second for a combination of alogliptin and pioglitazone) in July 2011.[11] In 2012, Takeda received a negative response from the USFDA on both of these NDAs, citing a need for additional data.[11]
In 2013 the FDA approved the drug in three formulations: As a stand-alone with the brand-name Nesina. Combined with metforminusing the name Kazano, and when combined with pioglitazone as Oseni.
Diabetes affects millions of people worldwide and is considered one of the main threats to human health in the 21st century. In 2006, the World Health Organization (WHO) estimated that over 180 million people worldwide had diabetes, and the number is projected to double by 2030. Over time, uncontrolled diabetes can damage body systems, including the heart, blood vessels, eyes, kidneys and nerves. According to the WHO, approximately 1.1 million people died from diabetes in 2005, and it is estimated that diabetes-related deaths will increase by more than 50% in the next decade. Globally, the socioeconomic burden of diabetes is substantial.
There are two main types of diabetes, designated type 1 and type 2, with type 2 diabetes accounting for over 90% of all diabetes cases globally. Type 1 diabetes is characterized by insulin deficiency, primarily caused by autoimmune-mediated destruction of pancreatic islet β-cells, and type 2 diabetes is characterized by abnormal insulin secretion and concomitant insulin resistance. To prevent the development of ketoacidosis, people with type 1 diabetes must take exogenous insulin for survival. Although those with type 2 diabetes are not dependent on exogenous insulin as much as subjects with type 1 diabetes, they may require exogenous insulin to control blood glucose levels.
As diabetes has become a global health concern, research interest in the condition has rapidly increased. In addition to studies on prevention, many studies with the aim of developing new interventions for the treatment of diabetes, especially type 2 diabetes, have been conducted. Currently available medications for the treatment and management of type 2 diabetes include metformin, sulfonylureas, thiazolidinediones and insulin. However, these therapies are commonly associated with secondary failure and may cause hypoglycemia. Insulin resistance and progressively worsening hyperglycemia caused by reduced β-cell function are major challenges in managing type 2 diabetes. Evidence suggests that patients with insulin resistance do not develop hyperglycemia until their β-cells are unable to produce enough insulin. New agents that can enhance insulin secretion from islet β-cells in a sustained glucose-dependent manner could therefore hold promise for the treatment of type 2 diabetes.
One promising approach is based on inhibition of the serine protease dipeptidyl- peptidase IV (DPP IV), a postproline dipeptidyl aminopeptidase that belongs to the S9b peptidase family of proteolytic enzymes. It is known that DPP IV plays a key role in maintaining glucose homeostasis by controlling the incretin activity of glucagon-like peptide 1 (GLP-I) and glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide). Inhibition of DPP IV is therefore recognized as a novel therapeutic approach for the treatment of type 2 diabetes.
Recently, a series of DPP IV inhibitors were developed. Among these highly potent compounds, alogliptin benzoate (SYR-322) and its analogs demonstrated encouraging antidiabetic efficacy (EP 1586571 (WO 2005/095381); WO 2008/067465; WO 2007/035379, and US 2004/097510).
Alogliptin benzoate can be prepared as described in EP 1586571 (WO 2005/095381) according to the process set forth in Scheme 1 :

Scheme 1
In accordance with this process, 6-Chlorouracil (1) is alkylated with 2- (bromomethyl)benzonitrile in the presence of NaH and LiBr in a mixture of DMF- DMSO to produce the TV-benzyluracil derivative (2) in 54% yield. Compound (2) is further alkylated with iodomethane and NaH in DMF/THF to give the 1 ,3 disubstituted uracil (3) in 72% yield. Subsequent displacement of chlorouracil (IV) with 3(R)- aminopiperidine dihydrochloride in the presence of either NaHCO3 in hot methanol or K2CO3 in aqueous isopropanol provides alogliptin (4), which is isolated as the corresponding benzoate salt by treatment with benzoic acid in ethanol. The overall yield of this three-stage process is -20-25%. One of the disadvantages of above described process is the difficulty to separate and purify mixtures of solvents with high boiling point (for example, DMF/DMSO) for recycling. Another disadvantage is the usage of hazardous materials such as sodium hydride, which requires anhydrous solvents as a reaction media.
Intermediate 2-((6-chloro-3-methyl-2,4-dioxo-3 ,4-dihydropyrimidin- 1 (2H)-yl)methyl) benzonitrile (3) is alternatively obtained by alkylation of 6-chloro-3 methyluracil with 2-(bromomethyl)benzonitrile by means of diisopropylethylamine in hot NMP (WO 2007/035629). Although this process is more technological than the previously described process (EP 1586571), the overall yield is still moderate (50-55%). The problem of mixed solvents (toluene, NMP, diisopropylethylamine) separation persists for this process as well.
………….
http://www.google.com/patents/EP2410855A1?cl=en
EXAMPLE 1
Preparation of (R)-2-((6-(3 -aminopiperidin-l-yl)-3 -methyl-2,4-dioxo-3 ,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 3):

Scheme 3
Preparation of l-(2-isocyanobenzyl)-3-methylurea
2-cyanobenzylamine hydrochloride (90 g) and Dichloromethane (800 ml) were taken into a round bottomed (RB) flask. Methyl isocyanate (45.6 g) was added at 5°C. Triethylamine (81 g) in Dichloromethane (300 ml) was added at the same temperature and stirred at room temperature for 16h. Water (1 L) was added and stirred for 30 min. The obtained solid was collected by filtration and dried in oven at 50°C for 12h. The yield is 85% and the purity 99.8%.
Preparation of l-(2-isocyanobenzyl)-3-methyIpyrimidine-2,4,6(lH,3H,5H)-trione
a). To a stirred solution of 0.11 mol of sodium ethanolate in 80 ml of ethanol abs. was added 0.1 mol of l-(2-isocyanobenzyl)-3-methylurea and 0.1 mol diethyl malonate. The mixture was refluxed for 3-5 h. The cooled residue was acidified with 0.1 M hydrochloric acid (60 ml). The solid which separated was filtered off and recrystallized from ethanol or any suitable solvent. The yield is 78-85% and purity >95%.
b). In an alternate embodiment, l-(2-isocyanobenzyl)-3-methylurea (30 g), acetic acid (105 ml) and malonic acid (18 g) were mixed and heated to 60°C. Acetic anhydride (60 ml) was added at 60°C and heating was continued for two hours at 80°C. The reaction mixture was poured over ice water (300 ml) and the obtained solid was filtered, washed with water (1×500 ml) and methyl-tert-butylether (100 ml). The yield is 60% with 93.4% purity.
The compound thus prepared can be used for the next step without purification or purified by crystallization or column chromatography.
Preparation of 6-chloro-l-(2-isocyanobenzyl)-3-methylpyriinidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4,6(lH,3H,5H)-trione (30 g) was mixed with phosphorus oxychloride (300 ml) and cooled to 0°C. Water (9 ml) was added slowly, stirred for 10 min. and heated to reflux at 110°C for 5h. Progress of the reaction was monitored by TLC (50% Ethyl acetate/Hexane). On completion of the reaction, phosphorus oxychloride was distilled off. The crude compound was dissolved in dichloromethane (500 ml) and poured into ice water (500 ml) by small portions. The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. The mixture of two isomers (4-chloro and 6-chloro derivatives = 1:1) was isolated and separated by column chromatography using neutral alumina and eluent – 25-50% of ethylacetate and hexane). The off-white solid was obtained, yield – 37%, purity – 99.8%. 1H NMR corresponds to literature data (J. Med. Chem. 2007, 50, 2297-2300).
b). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (18 mmol), phosphorus oxychloride (85 ml), benzyltriethylammonium chloride (16.5 g, 72 mmol) and phosphorus pentachloride (3.8 g, 18 mol) in acetonitrile (80 ml) was refluxed for 4-5 h with stirring. After evaporation under reduced pressure, the resulting oily residue was mixed with methylene chloride (or chloroform) and the mixture was poured into water and ice (50 ml). The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. Crude product was crystallized from THF-hexanes to give desired compound in 70.5% yield.
c). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (13.1 mmol) in POCl3 (30 ml) was refluxed for 1-3 h. The solvent was concentrated and then partitioned with CH2Cl2 (100 ml) and water (100 ml). The organic layer was washed with brine, dried over Na2SO4, and concentrated to give 6-chloro compound as a solid (-95%). Compound can be also precipitated from concentrated methylene chloride solution by hexanes and used as a crude for the next step or purified by reslurring in isopropanol, filtered off, washed with isopropanol, and dried under vacuum at 55-60° C.
Preparation of (R)-tert-butyl l-(3-(2-isocyanobenzyI)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate
a). 6-chloro- l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (13 g), Dimethylformamide (130 ml), Potassium carbonate (13 g) and tert-butyl (R)-piperidin- 3-ylcarbamate (10.4 g) were heated to 80°C for 7 hrs. The mixture was then allowed to come to room temperature and poured over ice water (500 ml). The obtained solid was filtered and washed with cold water (500 ml). The solid thus obtained was taken in Methyl-tert-butylether (50 ml) stirred for 10 min. filtered and washed with Hexane (50 ml), to give the N-tert-butyloxycarbonyl protected compound in -75% yield. b). In an alternate embodiment, a flask charged with a stir bar, 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (4.10 mmol), (Λ)-3- terrtnityloxycarbonylaminopiperidine (4.64 mmol), K2CO3 (1.15 g, 8.32 mmol) and DMF (12 mL) was stirred at 75 °C for 6 h. Then, water was added and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give the N-ter/butyloxycarbonyl protected compound in -93-96% yield.
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile salts
a). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile hydrochloride
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from previous procedure was dissolved in THF and acidified with 6M hydrochloric acid while maintaining the temperature below 15° C. The resultant slurry was cooled to 0-5° C, stirred at this temperature for 3-5 h and then filtered. The filter cake was washed twice with isopropanol and dried in vacuum at 45-5O0C to provide hydrochloride as a white crystalline solid.
b). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile trifluoroacetate
TFA (ImL) was added into the methylene chloride solution of (R)-tert-butyl l-(3-(2- isocyanobenzyl)- 1 -methyl-2,6-dioxo- 1 ,2,3,6-tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from the above-mentioned procedure. The solution was stirred at room temperature for 1 h and then the mixture was concentrated in vacuo. The residue was dissolved in a small amount of MeOH or isopropanol and the desired salt was precipitated by addition of diisopropyl ether. The solids were filtered off, washed with diisopropyl ether and dried in vacuum at 45-5O0C to provide trifluoroacetate as an off- white powder. c). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile benzoate (Alogliptin)
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate was dissolved in ethanol. A solution of benzoic acid in ethanol was added and the mixture was slowly heated to 65-70°C. The solution was stirred at this temperature for Ih and was then crystallized by cooling to 0-5° C and stirring for 12 hrs. The solution was filtered, washed with alcohol. The wet cake was then conditioned under nitrogen for 2 hours. The cake was dried for 8 hrs at 40-50° C to provide the benzoic acid salt of alogliptin as a white crystalline solid.
EXAMPLE 2:
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 4)


Scheme 4 Preparation of 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylurea (0.2 mol) and cyanoacetic acid (0.22 mol) were dissolved in acetic anhydride (400 ml), and the mixture was heated at 80°C for 2 hours. Acetic anhydride was distilled off under reduced pressure and water (200 ml) was added. The mixture was cooled to 0-5 0C and 2N NaOH solution (220 ml) was added and stirring was continued for 2 hours. The obtained solids were filtered off, washed with cold methanol and dried under vacuum. The yield of 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione was 72 %.
b). Under nitrogen atmosphere, l-(2-isocyanobenzyl)-3-methylurea (98.4 g) and cyanoacetic acid (80.0 g) was added to N,N-dimethylformamide (836 ml). The mixture was stirred at room temperature and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at this temperature. The mixture was stirred at room temperature for 4 hrs, cooled with water, and water-isopropanol [2:1 (volume ratio), 1670 ml] was added drop wise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 3-(2-cyano-acetyl)-3- methyl-l-(2-isocyanobenzyl)-urea with 68% yield.
To 3-(2-cyano-acetyl)-3-methyl-l-(2-isocyanobenzyl)-urea (120 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml), and the mixture was stirred with heating at 80° C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-l-(2-isocyanobenzyl)-3- methylpyrimidine-2,4(lH,3H)-dione in 76% yield.
c). 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (0.1 mol) was mixed with (R)-piperidin-3-yl-carbamic acid tert.-butyl ester hydrochloride (0.1 mol) of the appropriate amine hydrochloride and (R)-piperidin-3-yl-carbamic acid tert.-butyl ester (0.1 mol). The mixture was heated at 100°C and bubbling continued for 3 hr. Water was added to the cooled mixture and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give N-tert-butyloxycarbonyl protected compound in ~93-96% yield.
d). Benzoate salt of alogliptin was prepared as described above. While certain embodiments of the invention have been illustrated and described, it will be clear that the invention is not limited to the embodiments described herein. Numerous modifications, changes, variations, substitutions and equivalents will be apparent to those skilled in the art without departing from the spirit and scope of the present invention as described by the claims, which follow.
………………
Patent EP2410855A1
http://www.google.com/patents/EP2410855A1?cl=en
…………..

NMR
SOURCE APEXBT


References
- “Takeda Submits New Drug Application for Alogliptin (SYR-322) in the U.S.” (Press release). Takeda Pharmaceutical Company. January 4, 2008. Retrieved January 9, 2008.
- Vipidia: EPAR summary for the public (European Medicines Agency)
- Feng, Jun; Zhang, Zhiyuan; Wallace, Michael B.; Stafford, Jeffrey A.; Kaldor, Stephen W.; Kassell, Daniel B.; Navre, Marc; Shi, Lihong; Skene, Robert J.; Asakawa, Tomoko; Takeuchi, Koji; Xu, Rongda; Webb, David R.; Gwaltney II, Stephen L. (2007). “Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV”. J. Med. Chem.50 (10): 2297–2300.doi:10.1021/jm070104l.PMID 17441705.
- “www.aace.com” (PDF).
- http://www.takeda.com/news/2013/20130618_5841.html
- Seino, Yutaka; Fujita, Tetsuya; Hiroi, Shinzo; Hirayama, Masashi; Kaku, Kohei (September 2011), “Efficacy and safety of alogliptin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, dose-ranging comparison with placebo, followed by a long-term extension study (abstract only)”, Current Medical Research and Opinion 27 (9): 1781–1792,doi:10.1185/03007995.2011.599371,PMID 21806314, retrieved April 26,2012
- Kutoh, Eiji; Ukai, Yasuhiro (2012),“Alogliptin as an initial therapy in patients with newly diagnosed, drug naïve type 2 diabetes: a randomized, control trial (abstract only)”, Endocrine(January 17, 2012), doi:10.1007/s12020-012-9596-0, PMID 22249941, retrieved April 26, 2012
- Bosi, Emanuele; Ellis, G.C.; Wilson, C.A.; Fleck, P.R. (October 2011), “Alogliptin as a third oral antidiabetic drug in patients with type 2 diabetes and inadequate glycaemic control on metformin and pioglitazone: a 52-week, randomized, double-blind, active-controlled, parallel-group study”, Diabetes, Obesity and Metabolism (October 27, 2011) 13 (12): 1088–1096, doi:10.1111/j.1463-1326.2011.01463.x, retrieved April 26,2012
- White WB, Cannon CP, Heller SR et al. (October 2013). “Alogliptin after acute coronary syndrome in patients with type 2 diabetes”. N. Engl. J. Med. 369(14): 1327–35.doi:10.1056/NEJMoa1305889.PMID 23992602.
- White WB, Zannad F (January 2014). “Saxagliptin, alogliptin, and cardiovascular outcomes”. N. Engl. J. Med. 370 (5): 484.doi:10.1056/NEJMc1313880.PMID 24482824.
- Grogan, Kevin (April 26, 2012),“FDA wants yet more data on Takeda diabetes drug alogliptin”,PharmaTimes (PharmaTimes), PharmaTimes online, retrieved April 26,2012
- “GEN News Highlights: Takeda Pulls MAA for Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. June 4, 2009.
| EP1083172A1 * | May 26, 1998 | Mar 14, 2001 | Rimma Iliinichna Ashkinazi | N-substituted derivatives of 5-oxyiminobarbituric acid |
| US2598936 * | Apr 13, 1950 | Jun 3, 1952 | Searle & Co | Disubstituted cyanoalkanoylureas and thioureas and methods for their production |
| US6066641 * | Dec 12, 1995 | May 23, 2000 | Euro-Celtique S.A. | Aryl thioxanthines |
| US6248746 * | Jan 7, 1999 | Jun 19, 2001 | Euro-Celtique S.A. | 3-(arylalkyl) xanthines |
| US20080194593 * | Jan 11, 2008 | Aug 14, 2008 | Rao Kalla | A2b adenosine receptor antagonists |
| WO1994003456A1 * | Aug 5, 1993 | Feb 17, 1994 | Boehringer Ingelheim Kg | Asymmetrically substituted xanthine with adenosine-antagonistic properties |
| WO2001029010A1 * | Oct 18, 2000 | Apr 26, 2001 | Amjad Ali | Gram-positive selective antibacterial compounds, compositions containing such compounds and methods of treatment |
| WO2007035629A2 * | Sep 15, 2006 | Mar 29, 2007 | Takeda Pharmaceutical | Process for the preparation of pyrimidinedione derivatives |
| WO2007150011A2 * | Jun 22, 2007 | Dec 27, 2007 | Smithkline Beecham Corp | Prolyl hydroxylase inhibitors |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
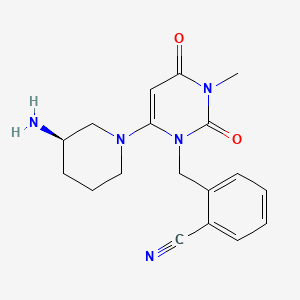
2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile
|
|
| Clinical data | |
| Trade names | Nesina, Vipidia Kazano, Vipidomet (withmetformin) Oseni, Incresync (withpioglitazone) |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | 20% |
| Metabolism | Limited, hepatic (CYP2D6– and3A4-mediated) |
| Biological half-life | 12–21 hours |
| Excretion | Renal (major) and fecal (minor) |
| Identifiers | |
| CAS Registry Number | 850649-62-6 |
| ATC code | A10BH04 |
| PubChem | CID: 11450633 |
| IUPHAR/BPS | 6319 |
| ChemSpider | 9625485 |
| UNII | JHC049LO86 |
| KEGG | D06553 |
| ChEBI | CHEBI:72323 |
| ChEMBL | CHEMBL376359 |
| Synonyms | SYR-322 |
| Chemical data | |
| Formula | C18H21N5O2 |
| Molecular mass | 339.39 g/mol |
 Alogliptin benzoate
Alogliptin benzoate
| MF: | C18H21N5O2.C7H6O2 |
| MW: | 461.519 |
| Melting Point: | 185-188°C |
| Optical Rotation: | -56.3° (c=1, MeOH) |
Solubility:Soluble in MeOH; Insoluble in ACN
850649-62-6 CAS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy


Sorry, the comment form is closed at this time.