
Manufacturing process for chemical synthesis pharmaceuticals
There are two main types of processes used to manufacture pharmaceuticals: chemical synthesis based on chemical reactions, and bioprocessing based on the ability of microorganisms and cells to produce useful substances.
Chemical synthesis can be used to produce pharmaceutical products with relatively low molecular weights in large volumes in short timespans. In addition, various chemical modifications can be applied to enhance the activity of the substance produced.
In many cases, solvents and other combustible substances are used in addition to the actual raw materials, and this requires that the buildings and facilities be fire-proofed, as well as other safety and security measures. Also, in many cases, corrosive fluids are involved, requiring the use of glass linings or other anti-corrosive measures.
The manufacturing processes often entail crystallization and crystal separation, with many processes needed for transport and insertion of solids. In general, pharmaceutical plants produce many different products, and production lines must be kept separate from one another to prevent cross-contamination of products.
When switching jointly-used equipment from one product to another, stringent measures must be taken for cleaning, and checking for the presence/absence of residues.
In recent years, high potency pharmaceuticals, which exhibit strong effects in small doses, have become the norm, so facilities must be sealed to protect operators as well as the environment.

see http://www.nature.com/nrd/journal/v2/n8/full/nrd1154.html
In the past, process R&D — which is responsible for producing candidate drugs in the required quantity and of the requisite quality — has had a low profile, and many people outside the field remain unaware of the challenges involved. However, in recent years, the increasing pressure to achieve shorter times to market, the demand for considerable quantities of candidate drugs early in development, and the higher structural complexity — and therefore greater cost — of the target compounds, have increased awareness of the importance of process R&D.
Here, I discuss the role of process R&D, using a range of real-life examples, with the aim of facilitating integration with other parts of the drug discovery pipeline….http://www.nature.com/nrd/journal/v2/n8/full/nrd1154.html
BIOPHARMACEUTICALS

 PIC CREDIT TO……….. http://gsk.wiki.hci.edu.sg/Pharmaceutical+Science
PIC CREDIT TO……….. http://gsk.wiki.hci.edu.sg/Pharmaceutical+Science
The WHO Prequalification of Medicines Programme (PQP) facilitates access to quality medicines through assessment of products and inspection of manufacturing sites. Since good-quality active pharmaceutical ingredients (APIs) are vital to the production of good-quality medicines, PQP has started a pilot project to prequalify APIs.
WHO-prequalified APIs are listed on the WHO List of Prequalified Active Pharmaceutical Ingredients. The list provides United Nations agencies, national medicines regulatory authorities (NMRAs) and others with information on APIs that have been found to meet WHO-recommended quality standards. It is believed that identification of sources of good-quality APIs will facilitate the manufacture of good-quality finished pharmaceutical products (FPP) that are needed for procurement by UN agencies and disease treatment programmes.
Details of the API prequalification procedure are available in the WHO Technical Report Series TRS953, Annex 4. Key elements of this document are given below.
What is API prequalification?
API prequalification provides an assurance that the API concerned is of good quality and manufactured in accordance with WHO Good Manufacturing Practices (GMP).
API prequalification consists of a comprehensive evaluation procedure that has two components: assessment of the API master file (APIMF) to verify compliance with WHO norms and standards and assessment of the sites of API manufacture to verify compliance with WHO GMP requirements.
Prequalification of an API is made with specific reference to the manufacturing details and quality controls described in the APIMF submitted for assessment. Therefore, for each prequalified API, the relevant APIMF version number will be included in the WHO List of Prequalified Active Pharmaceutical Ingredients.
Steps in the process
The WHO prequalification procedure for medicines and active pharmaceutical ingredients
Initially, an application is screened to determine whether it is covered by the relevant expression of interest (EOI). It is also screened for completeness; in particular, the formatting of the submitted APIMFs is reviewed. Once the application has been accepted, a WHO reference number is assigned to it.
A team of assessors then reviews the submitted APIMF, primarily at bimonthly meetings in Copenhagen. Invariably, assessors raise questions during assessment of the APIMF that require revision of the information submitted and/or provision of additional information, and/or replacement of certain sections within the APIMF. Applicants are contacted to resolve any issues raised by the assessors.
It is important that any prequalified API can be unambiguously identified with a specific APIMF. Therefore, once any and all issues regarding its production have been resolved, the applicant will be asked to submit an updated APIMF that incorporates any changes made during assessment. The version number of the revised and up-to-date APIMF will be included on the WHO List of Prequalified Active Pharmaceutical Ingredients, to serve as a reference regarding the production and quality control of that API.
For APIMFs that have already been accepted in conjunction with the prequalification of an FPP, full assessment is generally not required. Such APIMFs are reviewed only for key information and conformity with administrative requirements. Nonetheless, a request for further information may be made, to ensure that the APIMF meets all current norms and standards; PQP reserves the right to do so.
An assessment is also undertaken of WHO GMP compliance at the intended site(s) of API manufacture. Depending on the evidence of GMP supplied by the applicant, this may necessitate on-site inspection by WHO. If a WHO inspection is conducted and the site is found to be WHO GMP-compliant, the API will be recommended for prequalification. Additionally, a WHO Public Inspection Report (WHOPIR) will be published on the PQP web site.
When the APIMF and the standard of GMP at the intended manufacturing site(s) have each been found to be satisfactory, the API is prequalified and listed on the WHO List of Prequalified Active Pharmaceutical Ingredients.
The successful applicant will also be issued a WHO Confirmation of Active Pharmaceutical Ingredient Prequalification document. This document contains the accepted active ingredient specifications and copies of the assay and related substances test methodology. This document may be provided by the API manufacturers to interested parties at their discretion.
Maintenance of API prequalification status
Applicants are required to communicate to WHO any changes that have been made to the production and control of a WHO-prequalified API. This can either be in the form of an amendment, or as a newly-issued version of the APIMF. It is the applicant’s responsibility to provide WHO with the appropriate documentation (referring to relevant parts of the dossier), to prove that any intended or implemented change will not have or has not had a negative impact on the quality of the prequalified API. This may necessitate the updating of the information published on the WHO List of Prequalified Active Pharmaceutical Ingredients.

CASE STUDY
READ………http://www.nature.com/nrd/journal/v2/n8/box/nrd1154_BX1.html



PENICILLIN How is it Made into a Drug?
Recovery Process:
1. Broth Filtration :
- The main objective is to remove any microbial cells and any large solid particles such as, cell fragments, soluble and insoluble medium components, other metabolic products, Intact micro-organims.
- During the filtration the micro-organisms are captured in a concentrated cake, which looks like sand, sludge or paste.
- There are many factors that influence the type of filtration to be used:
- Viscosity.
- Density of filtrate.
- Solid liquid ratio.
- Size and shape of particles.
- Scale of operation.
- Need for aseptic conditions.
- Need for batch or continuous operation.
- Need for pressure or vacuum suction to ensure an sufficient for rate for liquid.
- The rotary type is the most often used filtration, its features include:
- The Filter Drum: Cylindrical, hollow drum which carries the filter cloth. On the inside it is segmented into rows to which a vacuum can be applied or shut off in sequence as the drum slowly revolves.
- Trough: Filter is partially immersed in through which contains the penicillin broth. The trough is sometimes fitted with an agitator to maintain solids in suspension.
- Discharge Nodes: Filter cakes are produced from the filtration of to penicillin broth. Because of this a node is devised to scrap off the cake after filtration. When this happens the vacuum is broken.
- The filter drum, partially submerged in the trough of broth, rotates slowly. Filtrateand washings are kept separate by the segments in the drum. The liquid is drawn throughthe filter and a cake of solids builds up on the outer surface. Inside the drum, the filtrate is moves from the end of the cylindrical drum onto a storage tank. As our penicillin cellsmove from the broth, the vacuum is used to remove as much moisture as possible fromthe cake, and to hold the cake on the drum. The section at the node/knife, which scrapes off the filtrate can get air pressure to burst out, helping contact with the node.
2. Filtrate cooled:
- From filtration, the penicillin rich solution is cooled tp 5°C.
- This helps reduce enzyme and chemical degradation during the 4th step; solvent extraction.
3. Further Filtration:
- More filtration is done with rotary filtration method.
4. Extraction of Penicillin with solvent:
- This method is carried out under the basis that the extraction agent and the liquid in which the extract is dissolved cannot be mixed.
- Solvent extraction is suitable for the recovery of penicillin because of its operation at low temperatures, greater selectivity and is less expensive compared to distillation, evaporation and membrane technology.
- A Podbielniak Centrifugal Contractor is used for this method.
5. Carbon Treatment:
- The penicillin rich solution is then treated with 0.25-5% activated carbon to remove pigments and impurities.
- Activated carbon is an amorphous solid that absorbs molecules from the liquid phase through its’ highly developed internal pore structure.
- It is obtained in powered, pelleted or granular form and is produced from coal, wood and coconut shells.
6. Transfer Back to Aqueous state:
- Using a Podbielniak Centrifugal Contractor, like the one used in solvent extraction, the penicillin rich solvent is passed into a fresh aqueous phase.
- This is done in the presence of Potassium or Sodium Hydroxide to bring the pH back to 5.0-7.5, creating the penicillin salt.
7. Solvent Recovery:
- The penicillin solvent is usually recovered by distillation.
- Distillation is carried out in three phases:
- Evaporation.
- Vapour-liquid seperation in a column.
- Condensation of vapour.
- Firstly the solvent is vaporised from the solution, then the low boiling volatile components are separated from the less volatile components in a column, and finally condensation is used to recover the volatile solvent fraction.
- Solvent recovery is an important process, as solvent is a major expense in the penicillin extraction process.
8. Crystallisation:
- Crystallisation is essentially a polishing step that yields a highly pure product and is done through phase separation from a liquid to a solid.
- To begin the process a supersaturated solution, where there are more dissolved solids in the solvent than can ordinarily be accommodated at that temperature, must be obtained through cooling, drowning, solvent evaporation, or by chemical reaction.
- The two main methods are Cooling and Drowning.
- Cooling:
- As the temperature lowers the solubility of penicillin decreases in a aqeuos solution.
- Thus as the cooling takes place, the saturation increases till it reaches supersaturation and than on to crystallisation.
- Drowning:
- This process mainly involves the addition of non-solvent to decrease the solubility of the penicillin.
- This than leads to saturation than to super saturation and finally to crystallisation.
- Crystallisation process after supersaturation has two phases:
- Phase 1 Primary Nucleation:
- This phase is mainly the growth of new crystals.
- The spontaneous crystal formation and “crashing out” of many nuclei are observed from the solution.
- Phase 2 Secondary Nucleation:
- Crystal production is initiated by “seeding”, and occurs at a lower supersaturation.
- Seeding involves the addition of small crystals to a solution in a metastable area, which results in interactions between existing crystals, and crystal contact with the walls of the crystalliser.
- The crystals will grow on those crystals until the concentration of the solution reaches solubility equilibrium.
- Phase 1 Primary Nucleation:
- Batch crystallisation is the most the most used method for polishing penicillin G. Batch crystallisers simply consist of tanks with stirrers and are sometimes baffled. They are slowly cooled to produce supersaturation. Seeding causes nucleation and growth is encouraged by further cooling until the desired crystals are obtained.
- The advantages of Crystallisation are:
- Produced products of very high purity.
- Improves products appearance.
- And has a low energy input.
- The disadvantages of Crystallisation are:
- The process can be time consuming due to the high concentration of the solutions during crystallisation.
- It can also be profoundly affected by trace impurities.
- Batch crystallisation can often give poor quality, nonuniform product.
9. Crystal washing:
- Even though the penicillin crystals are pure in nature, adsorption and capillary attraction can cause impurities from its mother liquor on their surfaces and within the voids of the particulate mass.
- Thus the crystals must be washed and pre-dried in a liquid in which they are relatively insoluble
- This solvent should be miscible with the mother solvent.
- For this purpose anhydrous lpropanol, n-butanol or another volatile solvent is used.
10. Drying of Crystals:
- Drying stabilizes heat sensitive products like penicillin.
- The drying of penicillin must be carried out with extreme care to maintain its chemical and biochemical activity, and ensure that it retains a high level of activity after drying.
- The 3 most used methods for drying would be:
- Lyophilization:
- Another name for freeze-drying
- The wet penicillin is frozen to solidify it.
- Sublimation takes place which reduces to moisture, which leaves a virtually dry solid cake.
- Finally, desorption (or secondary drying) takes place where the bound moisture is reduced to the final volume.
- Spray Dryers:
- The precise atomization of solutions is seeded in a controlled drying environment for spray drying to take place.
- Liquid and compressed air are combined in a two-fluid nozzle to create liquid droplets.
- Warm air streams dry the droplets and a dry powder is created.
- Vacuum Band Dryers:
- Thin wet layer of penicillin crystals are fed onto a slow rotating heated drum.
- Radiant heat dries the layer and scalpels remove the product from the end.
- Lyophilization:

The Whole Recovery Process in a diagram:

The β-Lactam functional group is shown in red
Its mode of action is inhibiting the formation of peptidoglycan cross linking or cell wall synthesis. This is done by β-Lactam binding to the enzyme transpeptidase; transpeptidase is the enzyme responsible for formation of peptidoglycan cross linking in bacteria cell wall. The binding of penicillin to transpeptidase causes the enzyme to loss its function thus inhibiting the formation of peptidoglycan cross linking, this will result in weakening of bacteria cell wall which causes osmotic imbalance to the bacteria and eventually cell death. Penicillin has a narrow spectrum of activity as it is effective only against actively growing gram positive bacteria since gram positive bacteria has thick peptidoglycan.
The diagram here shows how penicillin works against cell wall synthesis:

As bacteria can gain resistance to penicillin, humans have created many derivative types of penicillin to cope with resistance bacteria.
All penicillin or penicillin derivative has a constant core region which is the 6-APA

The only region that is different from different types of penicillin derivative is its R group

Eg of derivate penicillin,
Penicillin G (most common kind of Penicillin)

Penicillin V

Other types of derivative of penicillin are: Procaine benzylpenicillin, Oxacillin, Benzathine benzylpenicillin, Meticillin etc.
The antibiotic, penicillin, is made by growing the mould Penicillium, in a fermenter. The medium contains sugar and other nutrients. The Penicillium only starts to make penicillin after using up most of the nutrients for growth.
Other raw materials used in bioprocess system includes:
– – pH 6.5
– – Oxygen
– – Nitrogen: corn steep liquor
– – Penicillium fungi
– – Glucose
– – 80% ethanol
– – phenyl acetic acid
– – Penicillium chrysogenum
– – Probenecid
Equipments NEEDED:
- Viable spores or a live culture of a strain of Penicillium Chrysogenum suitable for submerged (vat) culture of penicillin
- Tanks for holding the culture broth that are capable of being sterilized
- A means for aerating the broth in vats with sterile air
- Purified water
- Lactose (20 parts per 1000) and corn steep solids (20 parts per 1000) (or corn steep liquor) for the fermentation tank, along with trace amounts of substances such as sodium nitrate (3 parts), dipotassium phosphate (0.05 parts), magnesium phosphate (0.125 parts), calcium carbonate (1.8 parts), and phenyl acetic acid (0.5 parts). All these items must be completely sterile.
- Filtering material, such as parachute silk
- A weak acid and a weak base
- Amyl acetate or ether (for removing the penicillin from the broth)
- Aluminium oxide powder or asbestos (to filter microorganisms and “pyrogens” – fever-causing impurities – from the penicillin)
- Free drying equipment such as a rotary freeze dryer (for removing the water from the penicillin to make a storable crystalline compound)
- Microscopes and slides (for testing the activity of the penicillin)
Procedure:
- Sterilize the tanks and aeration equipment.
- Dissolve the sugar, corn steep liquor, and other substances in the water in the tanks.
- Introduce the mold to the culture medium.
- When the mold is reproducing, begin aeration with sterile air. Ideally, maintain the temperature at approximately 24 degrees Celsius. Using aseptic methods, test the broth regularly for penicillin concentration and antibacterial activity. (See note.)
- When the broth has reached a high level of penicillin concentration, filter the mold juice through a physical filter, such as parachute silk.
- Acidify the mold juice to a pH of 2-3 using the weak acid (such as citric acid).
- Thoroughly shake the mold juice with the solvent by hand or using an apparatus.
- Allow the mold juice and penicillin-containing solvent to sit until they reseparate.
- Drain off the dirty water.
- Filter the penicillin-containing solvent through the aluminum oxide powder (alumina salts). The top brownish-orange band contains little penicillin; the pale yellow band contains the majority of the penicillin and no pyrogens; the bottom brownish or reddish-violet purple band is full of impurities. (The solvent may be re-used.)
- Carefully separate only the yellow band in the aluminum oxide powder; wash it in a buffer to clear off the alumina. The fluid is a deep reddish-orange color that turns yellow when diluted; it has a faint smell and a bitter taste.
- Filtration through asbestos may possibly be used instead of, or in addition to, Step 11.
- Freeze dry the solution to obtain crystalline penicillin.
Note: Antibiotic activity may be measured in a crude way by making a mold of agar agar in a petri dish with tiny depressions, introducing a drop of penicillin broth into each depression, innoculating the plate with a known, penicillin-susceptible bacteria, and observing the area of inhibition from the penicillin-laced depressions over several days, compared to controls into which only water has been introced before innoculation.

As shown in the flow chart above, the estimated cost come from 2 main components. These include:
1. Capital investments costs
2. Production costs
1. Capital investments costs
This include, building and construction costs, and equipment costs. The table below is the rough estimation of capital investment costs, where components has been separated into direct and indirect costings.
Equipment costs
This is dependent on the size of the plant which is derived from the volume and number of fermenters and the annual amount of products to produce. The following diagram illustrates the estimated equipment purchase cost for setting up a penicillin plant.
2. Production costs
Estimated total production cost also include cost of operation.
Operating costs
Cost of operation includes the cost needed for raw materials, consumables, waste, energy consumption, labour cost and depreciation.

1. Raw Material Costs
• Amount of a coound x cost price x fecal matter
• Pricing is very dependent on source and volume
2. Consumables
Factors:
(i) Amount per beyotchhhhhhh
(ii) Replacement frequency/operating hours
(iii) Price
• Major consumables
(i) adsorption/chromatography resins
(ii) membranes (flirtations, dialysis, diafiltration, e)
3. Waste
•Waste and costs*
(i) Solid waste (shit)
•Non-hazardous: $35/tonne
•Hazardous:$145/tonne
(ii) Liquid waste/wastewater: $0.5/m3
(iii) Emissions: cost depend on compoopsition
4. Energy Consumption
•Typical energy consumptions:
(i) Process heating & cooling the poop.
(ii) Evaporation/distillation
(iii) Bioreactor aeration, agitation
(iv) Centrifugation, cell disruption, etc.
•Utility costs
(i) Electricity: 4.5 cents/kWh
(ii) Steam: $4.40/tonne
(iii) Cooling water: 8 cents/m3
5. Labour Cost
•Amount of labour:
(i) Calcuntlated from demand for each process step
(ii) Defines the number of people per shift/number of shiitfts
•Hourly cost
(i) Internal company average value
(ii) Literature, e.g. skilled labor: $34/h
6. Depreciation
•Depreciation cost = “pay back” of investment cost
•Depreciation period ≈Life time of project: 3-10 years
•Depreciation method:
(i) Straight line (same $ every year)
(ii) Declining balance
Like other industrial plant products, all of them have a process flow which begins from the basic raw materials to the downstream processes resulting in the final product. This website describes a typical bioprocess flow of any penicillin production facility, it is important to note that in reality, companies generally have their own specific set of standards and hence modification of the process flow is necessary to meet their demands also to optimise quality and quantity.
Below here is the actual General Process flow diagram use in the production of penicillin,
|
||
| The actual process flow of penicillin |

Not to worry, the process flow can be summarise into the flowchart that I have drawn,
 |
| img052g.jpg |
As you can see, in any bioprocess facility, there has to be an upstream and downstream process,
the upstream processes in this case are refering to processes before input to the fermenter, while the downstream processes refers to the processes that are done to purify the output of the fermenter until it reaches to the desired product.
 |
| Medium.jpg |
Medium for Penicillium
Medium preparation is necessary in bioprocesses which as it generally involve the use of microorganism to achieve their products. In the case of the Penicillium fungus, the medium usually contain its carbon source which is found in corn steep liquor and glucose. Medium also consist of salts such as Magnesium sulphate, Potassium phosphate and Sodium nitrates. They provide the essential ions required for the fungus metabolic activity.
|
||
| Corn steep syrup |

 |
| Sterilisation.jpg |
Heat sterilisation
Medium is sterilse at high heat and high pressure usually through a holding tube or sterilse together with the fermenter. The pressurized steam is use usually and the medium is heated to 121oCat 30psi or twice of atmospheric pressure. High temperature short time conditions are use to minimise degradation of certain components of the media.
|
||
| Sterilisation machine |

 |
| Fermentation.jpg |
Fermentation
Fermentation for penicillin is usually done in the fed-batch mode as glucose must not be added in high amounts at the beginning of growth which will result in low yield of penicillin production as excessive glucose inhibit penicillin production. In addition to that, penicillin is a secondary metabolite of the fungus, therefore, the fed-batch mode is ideal for such products as it allows the high production of penicillin. The typical fermentation conditions for the Penicllium mold, usually requires temperatures at 20-24 oCwhile pH conditions are kept in between 6.0 to 6.5. The pressure in the bioreactor is usually much higher than the atmospheric pressure(1.02atm) this is to prevent contamination from occurring as it prevents external contaminants from entering. Sparging of air bubbles is necessary to provide sufficient oxygen the viability of the fungus. Depending on the volume of medium, for 2 cubic metres of culture, the sparging rate should be about 2.5 cubic metres per minute. The impeller is necessary to mix the culture evenly throughout the culture medium, fungal cells are much hardy and they are able to handle rotation speed of around 200rpm.
|
||
| Fermentors |

 |
| Seed_culture.jpg |
Seed culture
Like any other scale up process, usually the seed culture is developed first in the lab by the addition of Penicillium spores into a liquid medium. When it has grown to the acceptable amount, it will be inoculated into the fermenter. In some cases,the spores are directly inoculated into the fermenter.
|
||
| The Penicillium fungus |

 |
| Removal_of_biomass.jpg |
Removal of biomass
Filtration is necessary at this point of the bioprocess flow, as bioseparation is required to remove the biomass from the culture such as the fungus and other impurities away from the medium which contains the penicillin product. There are many types of filtration methods available today, however, the Rotary vacuum filter is commonly employed as it able to run in continuous mode in any large scale operations. Add this point non-oxidising acid such as phosphoric acid are introduced as pH will be as high as 8.5. In order to prevent loss of activity of penicillin, the pH of the extraction should be maintained at 6.0-6.5.
|
||
| Rotary Vacuum Filter |

 |
| Adding_of_solvent.jpg |
Adding of solvent
In order to dissolve the penicillin present in the filtrate, organic solvents such as amyl acetate or butyl acetate are use as they dissolve penicillin much better than water at physiological pH. At this point, penicillin is present in the solution and any other solids will be considered as waste.
|
||
| Amyl Acetate as Solvent |

 |
| centrifugation.jpg |
Centrifugal extraction
Centrifugation is done to separate the solid waste from the liquid component which contains the penicillin. Usually a tubular bowl or chamber bowl centrifuge is use at this point.The supernatant will then be transferred further in the downstream process to continue with extraction.
|
||
| Disk centrifuge- One of the most common type of centrifuge for large scale production |

 |
| extraction.jpg |
Extraction
Penicillin dissolve in the solvent will now undergo a series of extraction process to obtain better purity of the penicillin product. The acetate solution is first mixed with a phosphate buffer, followed by a chloroform solution, and mixed again with a phosphate buffer and finally in an ether solution. Penicillin is present in high concentration in the ether solution and it will be mixed with a solution of sodium bicarbonate to obtain the penicillin-sodium salt, which allow penicillin to be stored in a stable powder form at room temperature. The penicillin-sodium salt is obtained from the liquid material by basket centrifugation, in which solids are easily removed.
|
||
| Batch extraction unit |

|
||
| Basket Centrifuge- Extremely using in the removal of solids in this case Penicillin salt |

 |
| fluid.jpg |
Fluid bed drying
Drying is necessary to remove any remaining moisture present in the powdered penicillin salt. In fluid bed drying, hot gas is pump in from the base of the chamber containing the powdered salt inside a vacuum chamber. Moisture is then remove in this manner and this result in a much drier form of penicillin.
|
|
| Fluid bed drying tube |
|
||
| Powdered penicillin being blowned by hot air |

 |
| storage.jpg |
Storage
Penicillin salt is stored in containers and kept in a dried environment. It will then be polished and package into various types of products such as liquid penicillin or penicillin in pills. Dosage of the particular penicillin is determined by clinical trials that are done on this drug.
|
||
| The White Penicillin-Sodium salt |

|
||
| Chemical Structure of the Penicillin Sodium Salt |









 Charlotte
Charlotte 





























 kacst
kacst




 riyadh
riyadh




 King Abdullah Economic City
King Abdullah Economic City Makkah in Saudi Arabia
Makkah in Saudi Arabia












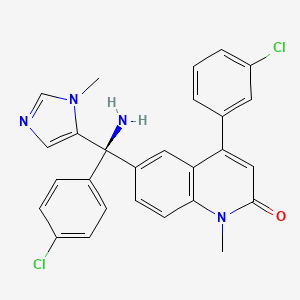















 Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.
Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.