












CMI 977



(2S,5S)-1-[4-[5-(4-Fluorophenoxymethyl)tetrahydrofuran-2-yl]-3-butynyl]-1-hydroxyurea 175212-04-1 CMI-977 is a potent 5-lipoxygenase inhibitor that intervenes in the production of leukotrienes and is presently being developed for the treatment of chronic asthma. It is a single enantiomer with an all–trans (2S,5S) configuration. Of the four isomers of CMI-977, the S,Sisomer was found to have the best biological activity and was selected for further development. The enantiomerically pure product was synthesized on a 2-kg scale from (S)-(+)-hydroxymethyl-γ-butyrolactone.
CytoMed, Inc. announced y the initiation of Phase I clinical trials for CMI-977, its orally active therapeutic product for the treatment of asthma. CMI-977 inhibits the 5-lipoxygenase (5-LO) cellular inflammation pathway to block the generation of leukotrienes, which play a key role in triggering bronchial asthma. The Company also announced that it has received a U.S. patent covering a number of 5-LO inhibitor compounds, including CMI-977, and their use in treating inflammatory and other disorders.
"Asthma is a chronic, persistent inflammatory disease of the airways characterized by coughing and wheezing. These symptoms are induced by the release of inflammatory mediators, including leukotrienes, from inflammatory cells in the lining of the airways," said Colin Scott, Vice President, Clinical and Regulatory Affairs of CytoMed. "CMI-977 inhibits the production of all classes of leukotrienes by inhibiting the 5-LO pathway. Preclinical studies of CMI-977 have shown similar efficacy to steroid treatment in reducing inflammation, without any evidence of the significant toxicity that has been associated with long-term use of steroids."
"CytoMed's product development strategy focuses on leveraging its expertise in molecular biology, medicinal chemistry and pharmacology to develop a broad range of product candidates," commented Thomas R. Beck, M.D., Chairman and CEO of CytoMed. "Moving our second product into the clinic is a significant step towards the Company's goal of developing a portfolio of safe and efficacious anti-inflammatory compounds." The Company's lead product, CMI-392, is currently in Phase II studies in collaboration with Stiefel Laboratories as a topical treatment for inflammation-related skin disorders.
The Phase I trial of CMI-977, which involves 56 healthy human volunteers, is being conducted at a single site. The double blind, randomized, escalating single dose study is designed to assess CMI-977's safety and tolerability.
The Company plans to complete the study in mid-1998. Over 14.6 million Americans suffer from chronic asthma. The disease is characterized by a widespread narrowing of the airways due to a contraction (spasm) of smooth muscle and overproduction of mucous, which blocks the air passages. These changes are caused by the release of spasmogens and vasoactive substances, including leukotrienes. Current long-term therapies include corticosteroids, which function by non-selectively suppressing a variety of cellular pathways that initiate inflammation. Steroids, while often effective, are associated with significant adverse side effects. CMI- 977 is a leukotriene modulator, part of a new class of drugs designed to
provide patients with a viable alternative to steroids.
CytoMed, Inc. is a growing biopharmaceutical company committed to the discovery and development of novel proprietary products for the treatment of inflammatory disease. The Company has three products in clinical or preclinical stage of development: CMI-392 in Phase II studies for the treatment of inflammatory skin disorders in collaboration with Stiefel
Laboratories; CMI-977, an orally active product in Phase I clinical trials for the treatment of asthma; and CMI-CAB-2, in late-stage preclinical development for the treatment of acute pulmonary and cardiovascular inflammation. To date, the Company has been funded primarily by investments from institutional and venture investors including Schroder Ventures, Oracle Strategic Partners, Atlas Venture, CIP Capital, BioAsia Investors, WPG Farber, Gateway Ventures, HealthCare Ventures and New York Life Insurance.
Org. Proc. Res. Dev., 1999, 3 (1), pp 73–76http://pubs.acs.org/doi/abs/10.1021/op980209l
………………………… 
PAPER
 A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.
A practical gram scale asymmetric synthesis of CMI-977 is described. A tandem double elimination of an α-chlorooxirane and concomitant intramolecular nucleophilic substitution was used as the key step. Jacobsen hydrolytic kinetic resolution and Sharpless asymmetric epoxidation protocols were applied for the execution of the synthesis of the key chiral building block.

http://www.sciencedirect.com/science/article/pii/S0957416603001575 ………………………………. 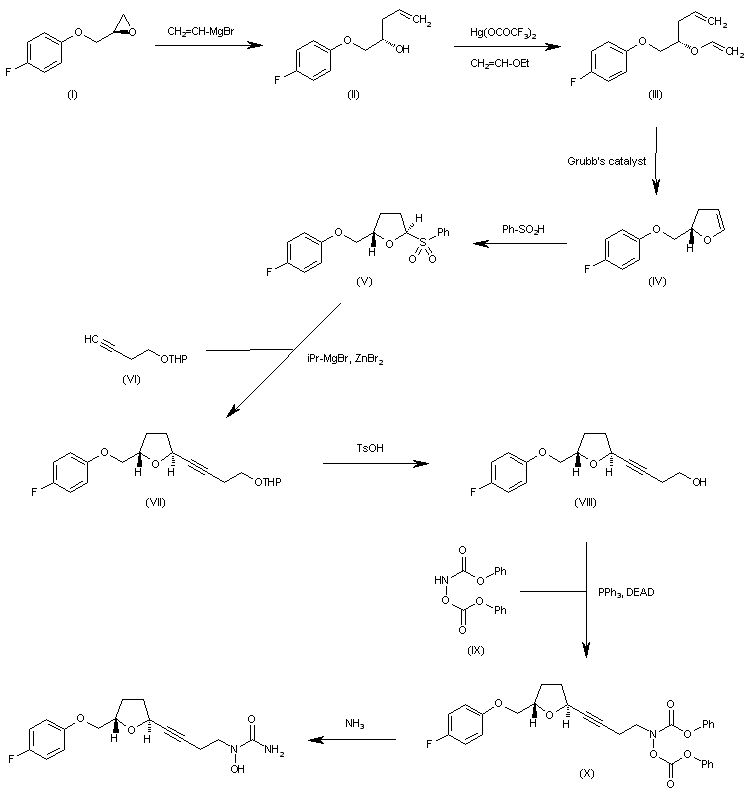 The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.http://www.chemdrug.com/databases/8_0_sluqxnnnfcuabcvj.html
The reaction of oxirane (I) with vinylmagnesium bromide in THF gives 1-(4-fluorophenoxy)-4-penten-2(S)-ol (II), which is treated with ethyl vinyl ether and mercuric trifluoroacetate to yield the vinyl ether (III). The cyclization of (III) by means of Grubb’s catalyst in refluxing benzene affords the dihydrofuran (IV), which is treated with benzenesulfinic acid in dichloromethane to give the sulfone (V). The reaction of (V) with the acetylenic tetrahydropyranyl ether (VI) by means of isopropylmagnesium bromide in THF yields the expected addition product (VII), which is treated with TsOH to eliminate the tetrahydropyranyl group and provide the alcohol (VIII). The condensation of (VIII) with N,O-bis (phenoxycarbonyl)hydroxylamine (IX) by means of PPh3 and DEAD in THF affords the protected carbamate derivative (X), which is finally treated with ammonia in methanol.http://www.chemdrug.com/databases/8_0_sluqxnnnfcuabcvj.html
””””””””””””””””””””
J. Braz. Chem. Soc. vol.24 no.2 São Paulo Feb. 2013
http://dx.doi.org/10.5935/0103-5053.20130024
http://www.scielo.br/scielo.php?pid=S0103-50532013000200003&script=sci_arttext Asthma is a chronic inflammatory disease of the respiratory system that results in the reduction or even the obstruction of air flow into the lungs.1 Over the last 40 years, there have been sharp increases in the global prevalence of asthma and the mortality due to this condition. In 2006, approximately 300 million people worldwide developed asthma, and there are approximately 180,000 deaths annually.2 In Brazil, asthma is the third most common cause of hospitalization in the Brazilian Unified Health System (SUS).3 The underdiagnosis and undertreatment of this disease have motivated the scientific community to search for new target-specific drugs to treat asthma and related respiratory diseases.4 The compound CMI-977 (LDP-977) (1) was discovered by Cyto-Med Inc., USA,5 and has been demonstrated to be a prominent candidate for the treatment of chronic asthma (Figure 1). This compound inhibits the 5-lipoxygenase pathway, thus blocking the production of leukotrienes.6 LDP-977 (1), containing a THF-2,5-trans-substituted ring with a (2S,5S) configuration, is orally active, and exhibits a good safety profile, a high degree of potency and excellent oral bioavailability relative to the three other stereoisomers.5

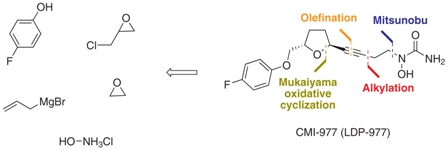
(2S,5S)-trans-5-[(4-Fluorophenoxy)methyl]-2-(4-N-hydroxyureidyl-1-butynyl)tetrahydrofuran, CMI-977 Over the years, several synthetic routes have been proposed for the stereoselective synthesis of the THF moiety present in CMI-977 (1) (Scheme 1).5,7,8  Intermediate 4 was prepared by Cyto-Med Inc., USA, using the first synthetic route developed,5 which involved a chiral pool approach for the creation of the C9 stereogenic center (Scheme 1). A nucleophilic attack involving an oxonium electrophile intermediate, obtained from 3, produced C6, but a disappointing low degree of selectivity was observed. In a similar oxonium strategy, Ley and co-workers7 employed an anomeric oxygen to promote the carbon rearrangement of an alkynyltributylstannane to access the THF unit, but their reaction also exhibited low selectivity (Scheme 1). Other similar strategies have led to similar results.8 Gurjar et al.9 reported a new stereoselective approach that installs the stereocenters at C6 and C9 in 6 using both Jacobsen hydrolytic kinetic resolution (HKR) and a Sharpless asymmetric epoxidation step (Scheme 1). The formation of a tandem propargyl alkoxide followed by intramolecular substitution resulted in the creation of the key tetrahydrofuran ring intermediate 7. Ley and co-workers10 also explored a similar tandem strategy providing the Retrosynthetic analysis of CMI-977 (LDP-977) (1) suitable intermediate 11, which in turn afforded the key fragment 7. These two new approaches were clearly Our disconnection approach began with a superior for the construction of the 2,5-anti THF unit as higher levels of diastereoselectivity were achieved. However, numerous steps are involved in these synthetic epoxide routes. In this paper, it is described our approach for the total synthesis of CMI-977 (LDP-977) (1). The biological importance of the target molecule and its structural features inspired us to devise a more concise and diastereoselective route to achieve the THF-2,5-trans ring of intermediate 7. Results and Discussion Retrosynthetic analysis of CMI-977 (LDP-977) (1) Our disconnection approach began with a long-established strategy for the insertion of the N-hydroxy urea moiety by alkylation involving acetylene 7 and epoxide 13, followed by a Mitsunobu-like reaction involving alcohol 4 and hydroxycarbamate 12 (Scheme 2).9,10 The terminal acetylene 7 can be assembled via Seyferth-Gilbert homologation (using the Ohira-Bestmann protocol)11 involving the aldehyde prepared from alcohol 14. It was intended to create the trans-THF configuration in our key fragment 14 using a Mukaiyama oxidative cyclization protocol with homoallylic alcohol 15.12 The functional groups in fragment 15 could be installed starting from commercially available and inexpensive 4-fluorophenol 16, rac-epichlorohydrin 17 and allylbromomagnesium 18, in a strategy similar to that applied by Gurjar et al.9 Preparation of the key fragment 14 Our approach to the total synthesis of CMI-977 (LDP-977) (1) began with the reaction of p-fluorophenol 16 with rac-epichlorohydrin 17 in the presence of KOH, providing rac-5 in 97% yield (Scheme 3).13
Intermediate 4 was prepared by Cyto-Med Inc., USA, using the first synthetic route developed,5 which involved a chiral pool approach for the creation of the C9 stereogenic center (Scheme 1). A nucleophilic attack involving an oxonium electrophile intermediate, obtained from 3, produced C6, but a disappointing low degree of selectivity was observed. In a similar oxonium strategy, Ley and co-workers7 employed an anomeric oxygen to promote the carbon rearrangement of an alkynyltributylstannane to access the THF unit, but their reaction also exhibited low selectivity (Scheme 1). Other similar strategies have led to similar results.8 Gurjar et al.9 reported a new stereoselective approach that installs the stereocenters at C6 and C9 in 6 using both Jacobsen hydrolytic kinetic resolution (HKR) and a Sharpless asymmetric epoxidation step (Scheme 1). The formation of a tandem propargyl alkoxide followed by intramolecular substitution resulted in the creation of the key tetrahydrofuran ring intermediate 7. Ley and co-workers10 also explored a similar tandem strategy providing the Retrosynthetic analysis of CMI-977 (LDP-977) (1) suitable intermediate 11, which in turn afforded the key fragment 7. These two new approaches were clearly Our disconnection approach began with a superior for the construction of the 2,5-anti THF unit as higher levels of diastereoselectivity were achieved. However, numerous steps are involved in these synthetic epoxide routes. In this paper, it is described our approach for the total synthesis of CMI-977 (LDP-977) (1). The biological importance of the target molecule and its structural features inspired us to devise a more concise and diastereoselective route to achieve the THF-2,5-trans ring of intermediate 7. Results and Discussion Retrosynthetic analysis of CMI-977 (LDP-977) (1) Our disconnection approach began with a long-established strategy for the insertion of the N-hydroxy urea moiety by alkylation involving acetylene 7 and epoxide 13, followed by a Mitsunobu-like reaction involving alcohol 4 and hydroxycarbamate 12 (Scheme 2).9,10 The terminal acetylene 7 can be assembled via Seyferth-Gilbert homologation (using the Ohira-Bestmann protocol)11 involving the aldehyde prepared from alcohol 14. It was intended to create the trans-THF configuration in our key fragment 14 using a Mukaiyama oxidative cyclization protocol with homoallylic alcohol 15.12 The functional groups in fragment 15 could be installed starting from commercially available and inexpensive 4-fluorophenol 16, rac-epichlorohydrin 17 and allylbromomagnesium 18, in a strategy similar to that applied by Gurjar et al.9 Preparation of the key fragment 14 Our approach to the total synthesis of CMI-977 (LDP-977) (1) began with the reaction of p-fluorophenol 16 with rac-epichlorohydrin 17 in the presence of KOH, providing rac-5 in 97% yield (Scheme 3).13  The epoxide rac-5was resolved by hydrolytic kinetic resolution under Jacobsen conditions,14 using the catalyst (R, R)-(salen)CoIII(OAc) (19, 0.5 mol%) and H2O (0.57 equiv) in tert-butyl methyl ether, providing (S)-5 in a 48% yield.9 The next step involved the epoxide ring-opening of (S)-5 with allylmagnesium bromide (18), providing homoallylic alcohol 15 in a quantitative yield (Scheme 4).
The epoxide rac-5was resolved by hydrolytic kinetic resolution under Jacobsen conditions,14 using the catalyst (R, R)-(salen)CoIII(OAc) (19, 0.5 mol%) and H2O (0.57 equiv) in tert-butyl methyl ether, providing (S)-5 in a 48% yield.9 The next step involved the epoxide ring-opening of (S)-5 with allylmagnesium bromide (18), providing homoallylic alcohol 15 in a quantitative yield (Scheme 4).  The subsequent oxidative cyclization of 15 according to the Mukaiyama protocol,12 mediated by the Co(modp)2 (20) (30 mol%) catalyst,15 provided trans-THF 14 as the only observed diastereoisomer in an 84% yield.8 This approach has proven to be a powerful strategy for accessing the 2,5-trans-THF unit in a highly diastereoselective fashion. Preparation of the key fragment 4 and conclusion of the synthesis The alcohol 14 was then oxidized to aldehyde 21 under Parikh-Doering conditions, followed by Seyferth-Gilbert homologation16 using the Ohira-Bestmann reagent 22,11 assembling the terminal acetylene 7 in a 75% yield over two steps (Scheme 5).
The subsequent oxidative cyclization of 15 according to the Mukaiyama protocol,12 mediated by the Co(modp)2 (20) (30 mol%) catalyst,15 provided trans-THF 14 as the only observed diastereoisomer in an 84% yield.8 This approach has proven to be a powerful strategy for accessing the 2,5-trans-THF unit in a highly diastereoselective fashion. Preparation of the key fragment 4 and conclusion of the synthesis The alcohol 14 was then oxidized to aldehyde 21 under Parikh-Doering conditions, followed by Seyferth-Gilbert homologation16 using the Ohira-Bestmann reagent 22,11 assembling the terminal acetylene 7 in a 75% yield over two steps (Scheme 5).  The 1H NMR and 13C NMR spectra and the optical rotation of trans-THF 7 matched the reported values for this compound.9 Next, the treatment of 7 with n-BuLi and ethylene oxide 13 led to alcohol 4 in a 70% yield. As shown in Scheme 5, the preparation of hydroxycarbamate 26 (53% yield), followed by its acetylation using acetyl chloride 27, provided 12 in a quantitative yield. A Mitsunobu-like reaction between alcohol 4 and N-hydroxycarbamate 12 provided 23 in a 93% yield. Finally, 23 was ammonolysed with NH3·MeOH, yielding CMI-977 as a white solid in a 38% yield. The spectral and physical data of the synthetic sample were in complete agreement with those reported in the literature.5,7-9
The 1H NMR and 13C NMR spectra and the optical rotation of trans-THF 7 matched the reported values for this compound.9 Next, the treatment of 7 with n-BuLi and ethylene oxide 13 led to alcohol 4 in a 70% yield. As shown in Scheme 5, the preparation of hydroxycarbamate 26 (53% yield), followed by its acetylation using acetyl chloride 27, provided 12 in a quantitative yield. A Mitsunobu-like reaction between alcohol 4 and N-hydroxycarbamate 12 provided 23 in a 93% yield. Finally, 23 was ammonolysed with NH3·MeOH, yielding CMI-977 as a white solid in a 38% yield. The spectral and physical data of the synthetic sample were in complete agreement with those reported in the literature.5,7-9
SPECTRAL DATA (2S,5S)-trans-5-[(4-Fluorophenoxy)methyl]-2-(4-N-hydroxyureidyl-1-butynyl)tetrahydrofuran, CMI-977 (1) To a round-bottomed flask, it was added 15 (85 mg, 0.19 mmol) at 0 ºC. Then, NH3 (2 mL, 14 mmol, 7 mol L-1in MeOH) was added, and the mixture was stirred at 0 ºC for 36 h. The reaction was concentrated under reduced pressure and purified by flash column chromatography using a mixture of CHCl3/MeOH (20:1) as the eluent, providing the compound CMI-977 (1) (24 mg, 0.074 mmol) as a colorless solid in a 38% yield; mp 106-107 ºC, 106-107 ºC;9
[α]D20 -40 (c 1.1, MeOH), [α]D -46.0 (c 1.1, MeOH);9
1H NMR (CDCl3, 250 MHz) δ 1.19 (s, 1H), 1.67-1.81 (m, 1H), 1.86-1.98 (m, 1H), 2.08-2.21 (m, 2H), 2.46 (t, 2H, J 6.5 Hz), 3.60 (t, 2H, J 6.8 Hz), 3.77-3.89 (m, 2H), 4.34-4.43 (m, 1H), 4.63-4.67 (m, 1H), 5.48 (s, 2H), 6.74-6.92 (m, 4H), 8.60 (br, 1H);
13C NMR (CDCl3, 150.9 MHz) δ 17.2 (CH2), 27.7 (CH2), 33.3 (CH2), 48.7 (CH2), 69.1 (CH), 70.7 (CH2), 76.9 (CH), 80.7 (C0), 82.9 (C0), 115.5 (CH), 115.7 (CH), 115.9 (CH), 154.8 (C0), 156.6 (C0), 158.2 (C0), 161.7 (C0);
IR (film) νmax/cm-1 3445, 3331, 3178, 2918, 2878, 1639, 1583, 1512, 1454, 1362, 1302, 1229, 1097, 1078, 1038, 937, 827, 762;
HRMS (ESI-TOF) m/z [M + H]+ for C16H20FN2O4 calcd. 323.1407, observed 323.1438.
References 1. Barnes P. J.; Br. J. Clin. Pharm. 1996,42, 3.
2. Braman, S. S.; Chest. 2006,130,4S. [ Links ]
3. Cabral, A. L. B.; Martins, M. A.; Carvalho, W. A. F.; Chinen,M.; Barbirotto, R. M.; Boueri, F. M. V.; Eur. Resp. J. 1998,12,35.
4. Jacobsen, J. R.; Choi, S. K.; Combs, J.; Fournier, E. J. L.; Klein, U.; Pfeiffer, J. W.; Thomas, G. R.; Yu, C.; Moran, E. J.; Bioorg. Med. Chem. Lett. 2012,22, 1213; [ Links ]
Millan, D. S.; Ballard, S. A.; Chunn, S.; Dybowski, J. A.; Fulton, C. K.; Glossop, P. A.; Guillabert, E.; Hewson, C. A.; Jones, R. M.; Lamb, D. J.; Napier, C. M.; Payne-Cook, T. A.; Renery, E. R.; Selby, M. D.; Tutt, M. F.; Yeadon, M.; Bioorg. Med. Chem. Lett.2011,21, 5826; [ Links ]
Sun, X. S.; Wasley, J. W. F.; Qiu, J; Blonder, J. P.; Stout, A. M.; Green, L. S.; Strong, S. A.; Colagiovanni, D. B.; Richards, J. P.; Mutka, S. C.; Chun, L.; Rosenthal, G. J.; ACS Med. Chem. Lett. 2011,2, 402; [ Links ]
Semko, C. M.; Chen, L.; Dressen, D. B.; Dreyer, M. L.; Dunn, W.; Farouz, F. S.; Freedman, S. B.; Holsztynska, E. J.; Jefferies, M.; Konradi, A. K.; Liao, A.; Lugar, J.; Mutter, L.; Pleiss, M. A.; Quinn, K. P.; Thompson, T.; Thorsett, E. D.; Vandevert, C.; Xu, Y.-Z.; Yednock, T. A.; Bioorg. Med. Chem. Lett .2011,21,1741. [ Links ]
5. Cai, X.; Hwang, S.; Killan, D.; Shen, T. Y.; US pat. 5,648,486 1997; [ Links ] Cai, X.; Grewal, G.; Hussion, S.; Fura, A.; Biftu, T.; US pat. 5,681,966 1997; [ Links ]
Cai, X.; Cheah, S.; Eckman, J.; Ellis, J.; Fisher, R.; Fura, A.; Grewal, G.; Hussion, S.; Ip, S.; Killian, D. B.; Garahan, L. L.; Lounsbury, H.; Qian, C.; Scannell, R. T.; Yaeger, D.; Wypij, D. M.; Yeh, C. G.; Young, M. A.; Yu, S.; Abs. Pap. Am. Chem. Soc.,1997,214,214-MEDI. [ Links ]
6. Cai, X.; Chorghade, M. S.; Fura, A.; Grewal, G. S.; Juaregui, K. A.; Lounsbury, H. A.; Scannell, R. T.; Yeh, C. G.; Young, M. A.; Yu, S.; Org. Process Res. Dev. 1999,3,73.
7. Dixon, D. J.; Ley, S. V.; Reynolds, D. J.; Chorghade, M. S.; Synth. Commun. 2000,30, 1955; [ Links ]Dixon, D. J.; Ley, S. V.; Reynolds, D. J.; Chorghade, M. S.; Indian J. Chem., Sect B 2001,40,1043.
8. Chorgade, M. S.; Gurjar, M. K.; Adikari, S. S.; Sadalapure, K.; Lalitha, S. V. S.; Murugaiah, A. M. S.; Radhakrishna, P.; Pure Appl. Chem. 1999,71, 1071; [ Links ] Gurjar, M. K.; Murali Krishna, L.; Sridhar Reddy, B.; Chorghade, M. S.; Synthesis 2000, 557; [ Links ] Chattopadhyay, A.; Vichare, P.; Dhotare, B.;Tetrahedron Lett. 2007,48,2871.
9. Gurjar, M. K.; Murugaiah, A. M. S.; Radhakrishna, P.; Ramana, C. V.; Chorghade, M. S.; Tetrahedron: Asymmetry 2003,14,1363.
10. Sharma, G. V. M.; Punna, S.; Prasad, T. R.; Krishna, P. R.; Chorghade, M. S.; Ley, S. V.; Tetrahedron: Asymmetry 2005,16,1113.
…………………………………………………
read
Pure Appl. Chem., Vol. 71, No. 6, pp. 1071-1074, 1999.
http://pac.iupac.org/publications/pac/pdf/1999/pdf/7106×1071.pdf
Full text – pdf 322 kB – IUPAC
………………………………………………… 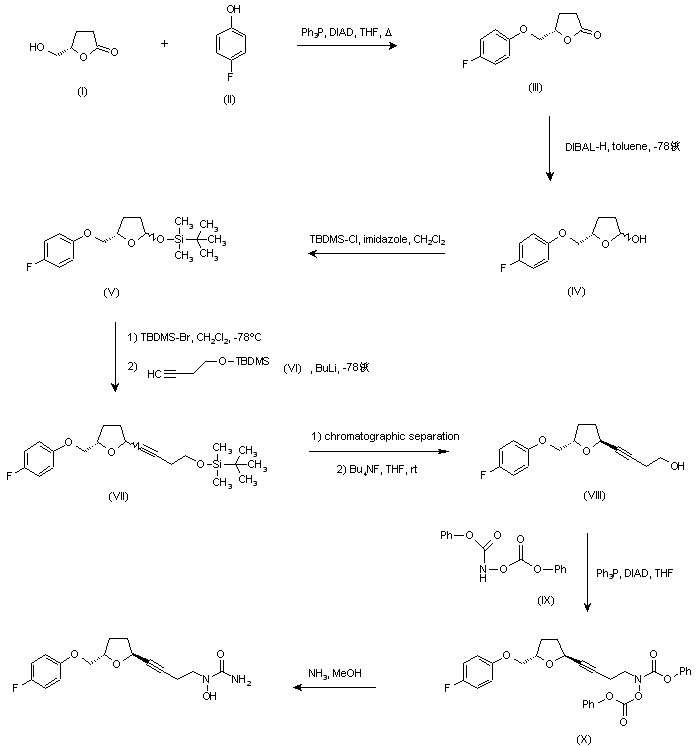 US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.http://www.chemdrug.com/databases/8_0_sluqxnnnfcuabcvj.html …………………………………………………. PAPER
US 5703093; US 5792776; WO 9600212 Ether (III) was prepared by condensation of (S)-4-(hydroxymethyl)butyrolactone (I) and 4-fluorophenol (II) in the presence of diisopropylazodicarboxylate (DIAD) and triphenylphosphine under Mitsunobu conditions. Then, reduction of lactone (III) with DIBAL-H in toluene at -78 C gave lactol (IV), which was converted to silyl ether (V) by treatment with tert-butyldimethylsilyl chloride (TBDMS-Cl) and imidazole. Subsequent reaction of (V) with TBDMS-Br in CH2Cl2 at -78 C, followed by condensation with the lithium acetylide derived from acetylene (VI), yielded compound (VII) as a mixture of isomers. Chromatographic separation of the mixture provided the desired trans isomer, which was deprotected by treatment with tetra-n-butylammonium fluoride to give alcohol (VIII). This was then condensed with N,O-bis(phenoxycarbonyl)hydroxylamine (IX) in the presence of DIAD and Ph3P to furnish the hydroxamic acid derivative (X). Finally, concomitant deprotection of the O-phenoxycarbonyl group and substitution of the remaining phenoxy group for an amino group by treatment with methanolic ammonia in a pressure tube, provided the title compound.http://www.chemdrug.com/databases/8_0_sluqxnnnfcuabcvj.html …………………………………………………. PAPER
| Title: | A short and efficient stereoselective synthesis of the potent 5-lipoxygenase inhibitor, CMI-977† |
| Authors: | Dixon, Darren J Ley, Steven V Reynolds, Dominic J Chorghade, Mukund S |
| Issue Date: | Nov-2001 |
| Publisher: | NISCAIR-CSIR, India |
| Abstract: | A short and efficient synthesis of the potent 5-lipoxygenase inhibitor CMI-977 has been accomplished, utilising an oxygen to carbon rearrangement of an anomerically linked alkynyl stannane tetrahydrofuranyl ether derivative as the key step. |
| Page(s): | 1043-1053 |
| CC License: |  CC Attribution-Noncommercial-No Derivative Works 2.5 India CC Attribution-Noncommercial-No Derivative Works 2.5 India |
| Source: | IJC-B Vol.40B(11) [November 2001] |
Files in This Item:
|
http://nopr.niscair.res.in/bitstream/123456789/22437/1/IJCB%2040B%2811%29%201043-1053.pdf ……………………………………………….
http://www.google.com.ar/patents/US20080081835 Specific inhibitors of 5-LO that may be mentioned include the following.
-
- (1) Zileuton (synonyms: A-64077, ABT 077, Zyflo®), described in, for example, EP 0 279 263, U.S. Pat. No. 4,873,259, Int. J. Immunopharmacol. 14, 505 (1992), Br. J. Cancer 74, 683 (1996) and Am. J. Resp. Critical Care Med. 157, Part 2, 1187 (1998).

-
- (2) A-63162, described in, for example, Anticancer Res. 14, 1951(1994).

-
- (3) A-72694.

-
- (4) A-78773, described in, for example, Curr. Opin. Invest. Drugs 2, 69 (1993).

-
- (5) A-79175 (the R-enantiomer of A 78773), described in, for example, Carcinogenesis 19, 1393 (1998) and J. Med. Chem. 40, 1955 (1997).

-
- (6) A-80263.

-
- (7) A-81834.

-
- (8) A-93178

-
- (9) A-121798, described in, for example, 211th Am. Chem. Soc. Meeting. 211: abstr. 246, 24 Mar. 1996.
- (10) Atreleuton (synonyms ABT-761 and A-85761), described in, for example, Exp. Opin. Therap. Patents 5 127 (1995).

-
- (11) MLN-977 (synonyms LPD-977 and CMI-977), described in, for example, Curr. Opin. Anti–Inflamm. &Immunomod. Invest. Drugs 1, 468 (1999). This, as well as similar compounds are described in U.S. Pat. No. 5,703,093.

…………………………………..
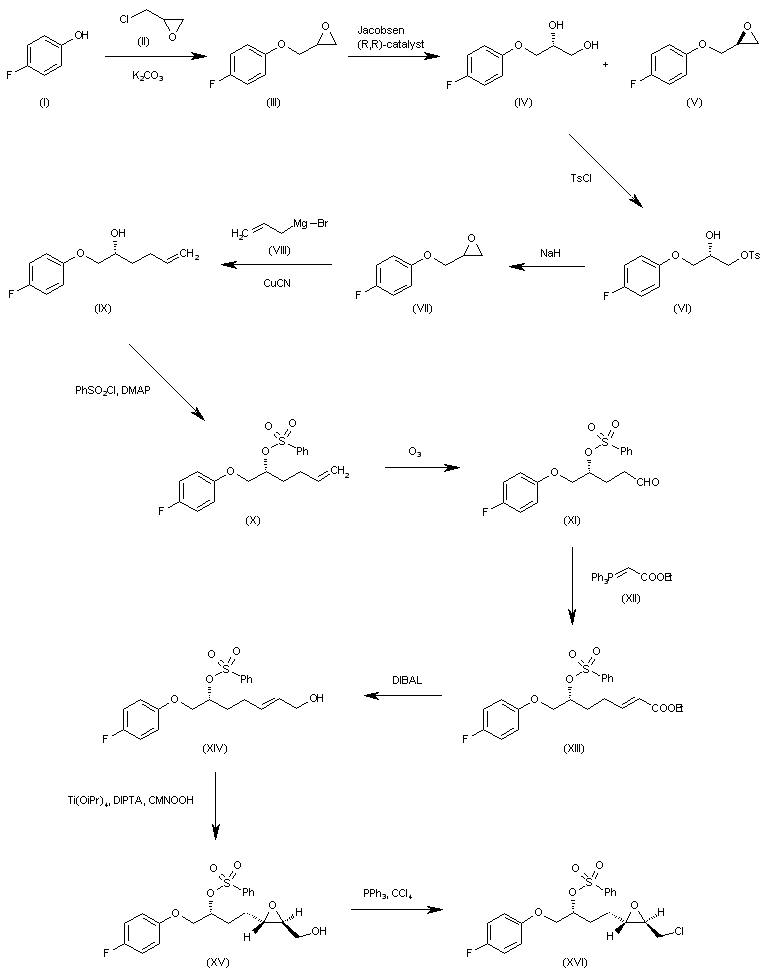 WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….
WO 0001381 The reaction of 4-fluorophenol (I) with epichlorohydrin (II) by means of K2CO3 in refluxing acetone gives 2-(4-fluorophenoxymethyl)oxirane (III), which is submitted to an enantioselective ring opening with the Jacobsen (R,R)-catalyst yielding a mixture of the (R)-diol (IV) and unaltered epoxide (V), easily separated by column chromatography. The reaction of (IV) with tosyl chloride and pyridine in dichloromethane affords the primary monotosylate (VI), which is converted into the chiral epoxide (VII) by reaction with NaH in THF/DMF. The reaction of (VII) with allylmagnesium bromide (VIII) in ethyl ether gives the 2-hexenol derivative (IX), which is treated with benzenesulfonyl chloride and DMAP yielding the sulfonate (X). The ozonolysis of (X) with ozone in dichloromethane affords the aldehyde (XI), which is condensed with ethoxycarbonylmethylene(triphenyl)phosphorane (XII) yielding the 2-heptenoic ester (XIII). The reduction of (XIII) with diisobutylaluminum hydride (DIBAL) in toluene/dichloromethane provides the 2-hepten-1-ol (XIV), which is epoxidized with cumene hydroperoxide in the presence of diisopropyl (+)-tartrate and Ti(Oi-Pr)4 in dichloromethane to give the chiral epoxyalcohol (XV). The reaction of (XV) with triphenylphosphine/CCl4 in chloroform affords the corresponding chloride (XVI). …………………………………….
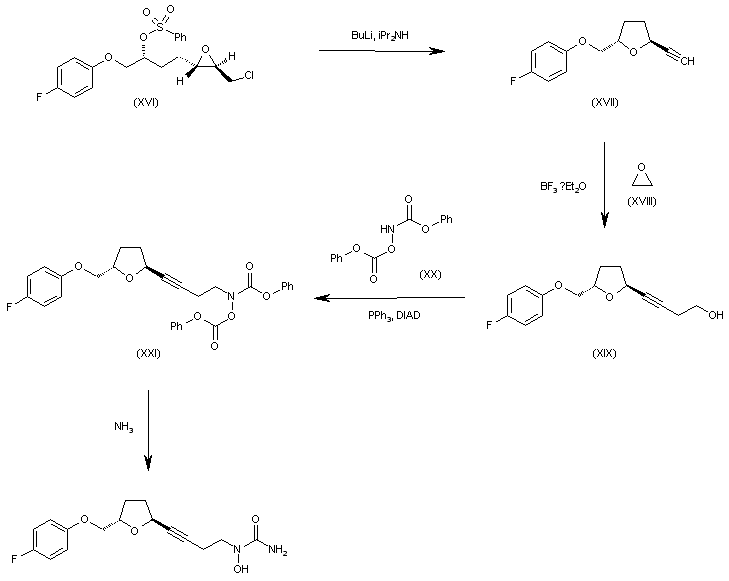 WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.
WO 0001381 Intermediate (XVI) is treated with BuLi and diisopropylamine in THF giving the chiral acetylenic tetrahydrofuran (XVII). The addition of ethylene oxide (XVIII) to the terminal acetylene of (XVII) by means of BF3/Et2O in THF gives the 3-butyl-1-ol derivative (XIX), which is condensed with N,O-bis(phenoxy- carbonyl)hydroxylamine (XX) by means of PPh3 and diisopropylazodicarboxylate (DIAD) in THF yielding the final intermediate (XXI). Finally, this compound is treated with ammonia in methanol to obtain the target urea derivative.
…………………………….
poster
http://www.prp.rei.unicamp.br/pibic/congressos/xxcongresso/paineis/092085.pdf
SÍNTESE TOTAL DO CMI-977 (LDP-977), UM PODEROSO AGENTE ANTIASMÁTICO
Lui Strambi Farina (IC), Marco Antonio Barbosa Ferreira (PG) e Luiz Carlos Dias (PQ)*
INSTITUTO DE QUÍMICA, UNIVERSIDADE ESTADUAL DE CAMPINAS, C.P. 6154, 13084-971, CAMPINAS, SP, BRASIL
*ldias@iqm.unicamp.br
Agência Financiadora: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ).
Palavras-Chave: Síntese orgânica, Tetrahidrofuranos, CMI-977 (LDP-977)
……………………………
Synthesis of (+)-Muricatacin and a Formal Synthesis of CMI-977 from l-Malic Acid
https://www.thieme-connect.de/DOI/DOI?10.1055/s-0033-1338934
A total synthesis of (+)-muricatacin and a formal synthesis of CMI-977 have been achieved using commercially available l-malic acid based on our furan approach to oxacyclic systems, the proven scope of which is thus broadened.

Sorry, the comment form is closed at this time.