











 |
|
| Systematic (IUPAC) name | |
|---|---|
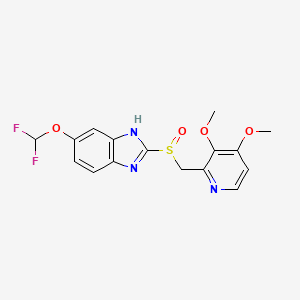
| (RS)-6-(Difluoromethoxy)-2-[(3,4-dimethoxypyridin-2-yl)methylsulfinyl]-1H-benzo[d]imidazole | |
| Clinical data | |
| Trade names | Protonix |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a601246 |
| Licence data | US FDA:link |
| Pregnancy cat. | B3 (AU) B (US) |
| Legal status | ℞ Prescription only |
| Routes | Oral and intravenous |
| Pharmacokinetic data | |
| Bioavailability | 77% |
| Metabolism | Hepatic (CYP3A4) |
| Half-life | 1 hour |
| Excretion | Renal |
| Identifiers | |
| CAS number | 102625-70-7 |
| ATC code | A02BC02 |
| PubChem | CID 4679 |
| DrugBank | DB00213 |
| ChemSpider | 4517 |
| UNII | D8TST4O562 |
| KEGG | D05353 |
| ChEBI | CHEBI:7915 |
| ChEMBL | CHEMBL1502 |
| Chemical data | |
| Formula | C16H15F2N3O4S |
| Mol. mass | 383.371 g/mol |
Pantoprazole is a proton pump inhibitor drug that inhibits gastric acid secretion.
Pantoprazole is a proton pump inhibitor drug used for short-term treatment of erosion and ulceration of the esophagus caused by gastroesophageal reflux disease.
Use
Pantoprazole is used for short-term treatment of erosion and ulceration of the oesophagus caused by gastroesophageal reflux disease. Initial treatment is generally of eight weeks’ duration, after which another eight week course of treatment may be considered if necessary. It can be used as a maintenance therapy for long term use after initial response is obtained.
Adverse effects
Antacid preparations such as pantoprazole work by suppressing the acid-mediated breakdown of proteins. This leads to an elevated risk of developing food and drug allergies due to undigested proteins passing into the gastrointestinal tract where sensitisation occurs. It is unclear whether this risk occurs with short-term or only long-term use.[1]
Common
- Gastrointestinal: Abdominal pain (3%), diarrhea (4%), flatulence (4%)
- Neurologic: Headache (5%)
Serious
- Gastrointestinal: Atrophic gastritis, clostridium difficile diarrhea
- Hematologic: Thrombocytopenia (less than 1%)
- Immunologic: Stevens-Johnson syndrome, toxic epidermal necrolysis
- Musculoskeletal: Muscle disorders, bone fracture and infection, Clostridium difficile, osteoporosis-related, hip fracture,rhabdomyolysis
- Renal: Interstitial nephritis (rare)
- Nutrition: May reduce the absorption of important nutrients, vitamins and minerals, as well as medications, leaving users at increased risk for pneumonia.[2]
- Cardiovascular: Increase in a chemical that suppresses the production of nitric oxide by 25% in humans, which have proven to relax and protect arteries and veins. Causes blood vessels to constrict, a development that could lead to a number of cardiovascular problems if continued for a prolonged period of time.[2]
Pharmacology

Wyeth pantoprazole 20mg.
Pantoprazole is metabolized in the liver by the cytochrome P450 system.[3] Metabolism mainly consists of demethylation by CYP2C19followed by sulfation. Another metabolic pathway is oxidation by CYP3A4. Pantoprazole metabolites are not thought to have any pharmacological significance. Pantoprazole is relatively free of drug interactions;[4] however, it may alter the absorption of other medications that depend on the amount of acid in the stomach, such as ketoconazole or digoxin. Generally inactive at acidic pH of stomach, thus it is usually given with a pro kinetic drug. Pantoprazole binds irreversibly to H+K+ATPase (proton pumps) and suppresses the secretion of acid. As it binds irreversibly to the pumps, new pumps have to be made before acid production can be resumed. The drug’s plasma half-life is about 2 hours.[5]
Pharmacokinetics
Absorption
- Bioavailability: (oral, delayed release tablets), approximately 77%
- Effect of food: (oral, delayed-release tablets), AUC and Cmax no effect, Tmax variable, absorption delayed, no net effect
- Effect of food: (oral, for-delayed-release suspension), administer 30 minutes before a meal
- Tmax, Oral, delayed-release suspension: 2 to 2.5 h
- Tmax, Oral, delayed-release tablets: 2.5 h
- Tmax, Oral, delayed-release tablets: 1.5 to 2 hours (pediatrics)
Distribution
- Protein binding: about 98% to primarily albumin
- Vd, extensive metabolizers (IV): approximately 11 L to 23.6 L
- Vd, pediatrics (oral): 0.21 to 0.43 L/kg.
Metabolism
- Hepatic; cytochrome P450 CYP2C19; minor metabolism from CYP3A4, 2D6, and 2C9
Excretion
- Fecal: (oral or IV, normal metabolizers), 18%
- Renal: (oral or IV, normal metabolizers), approximately 71%, none as unchanged
- Dialyzable: no (hemodialysis)
- Total body clearance: (IV) 7.6 to 14 L/hour.
- Total body clearance: (oral, pediatrics) 0.18 to 2.08 L/h/kg
Elimination Half Life
- Oral or IV, 1 hour
- Oral or IV, slow metabolizers, 3.5 to 10 hours
- Pediatrics, 0.7 to 5.34 hours
Availability
Pantoprazole was developed by Altana (owned by Nycomed) and was licensed in the USA to Wyeth (which was taken over by Pfizer). It was initially marketed under the brand name Protonix by Wyeth-Ayerst Laboratories and now is available as a generic. It is available by prescription in delayed-release tablets. It is also available for intravenous use.
On 24 December 2007, Teva Pharmaceutical released an AB-rated generic alternative to Protonix.[6] This was followed by generic equivalents from Sun Pharma and Kudco Pharma. Wyeth sued all three for patent infringement and launched its own generic version of Protonix with Nycomed.[7][8]
On October 18, 2010 the U.S. Food and Drug Administration (FDA) accepted the filing of an ANDA for a delayed release generic version of Protonix by Canadian companyIntelliPharmaCeutics.[9]
Brand names
Pantoprazole is available from a range of international suppliers under brand names including Pantazone, Pantop-D, Pantasan, Pantrol, Prazolin, Pantochem, Pansev, Pantec, Somac, API, Tecta, Protium, Pantodac, Perizole, Pansped, Percazole, Astropan, Fenix, Pantecta, Pantoloc, Controloc, Somac, Tecta, Protium, Inipomp, Eupantol, Pantozol, Pantodac, Perizole, Pansped, Zurcazol, Protonex, Pantup,Pantomed, TopZole, Nolpaza, Controloc, UXL-D, Pantid, Pantogen, Pantpas and Prazolin.
Pantoprazole sodium salt
The structural formula

Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | A02BC02 A02BD04 |
C 16 H 14 F 2 N 3 NaO 4 S | 405.36 g / mol | 138786-67-1 |
| hydrate | A02BC02 A02BD04 |
C 16 H 14 F 2 N 3 NaO 4 S · 3 / 2H 2 O | 864.76 g / mol | 164579-32-2 |
| (+) – Isomer | A02BC02 A02BD04 |
C 16 H 14 F 2 N 3 NaO 4 S | 405.36 g / mol | 160098-11-3 |
| (-) – Isomer | A02BC02 A02BD04 |
C 16 H 14 F 2 N 3 NaO 4 S | 405.36 g / mol | 160488-53-9 |
| racemate | A02BC02 A02BD04 |
C 16 H 14 F 2 N 3 NaO 4 S | 405.36 g / mol | 142678-34-0 |
Application
-
agent for the treatment of gastric ulcer
-
inhibitor of gastric H + / K + ATPase
Classes of substances
-
Benzimidazoles, 2 (alkylsulfinyl) benzimidazoles
-
Fluoro-ethers
-
Pyridines
-
-
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2428870 | 2006-05-23 | 2021-11-17 |
| Canada | 2092694 | 2005-04-05 | 2011-09-06 |
| Canada | 2341031 | 2006-04-04 | 2019-08-12 |
| United States | 7544370 | 2006-12-07 | 2026-12-07 |
| United States | 4758579 | 1993-07-19 | 2010-07-19 |
Synthesis pathway
| Synthesis a) |
|---|
      |

http://www.google.com/patents/EP1335913A1?cl=en
Pantoprazole is the international non-proprietary name of the chemical product 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2- pyridinyl)methyl]sulfmyl]-lH-benzimidazole of formula

Pantoprazole This product is an active ingredient used in the treatment of gastric ulcers, usually in the form of its sodium salt.
The product was described for the first time in European patent application EP-A-0166287 that also describes several processes for the preparation of products assignable to a general formula among which pantoprazole is to be found. The reaction sequences of these processes, applied precisely to the preparation of pantoprazole, are given in Scheme 1.

Scheme 1
In Scheme 1, the variables Y, Z, Z’ and Z” are leaving groups, for example atoms of halogen, and the variables M and M’ are atoms of alkali metals.
Austrian patent AT-B-394368 discloses another process based on a different route of synthetis, the reaction sequence of which is given in Scheme 2.

Pantoprazole Scheme 2
Nevertheless, this process has obvious drawbacks, since the methylation can take place not only in OH in the 4-position of the pyridine ring, but also in the nitrogen linked to a hydrogen of the benzimidazole ring, which can give place to mixtures of the desired product with the two possible methylated isomers of the benzimidazole compounds obtained, 3- methyl or 1 -methyl, which means that additional chromatographic purification steps are needed and the yields obtained are low.
PCT application WO97/29103 discloses another process for the preparation of pantoprazole, the reaction sequence of which is given in Scheme 3.

Scheme 3 As may be seen, different synthesis strategies have been proposed for the preparation of pantoprazole, some of them recently, which is an indication that the preparation of the product is still not considered to be sufficiently well developed, whereby there is still a need for developing alternative processes that allow pantoprazole to be prepared by means of simpler techniques and more accessible intermediate compounds and with good chemical yields.
EXAMPLES
Example 1. – Preparation of compound (IX)

47.5 ml (0.502 mol) of acetic anhydride were mixed with 1.65 g (0.0135 mol) of 4-dimethylaminopyridine, giving a transparent yellow solution which was heated to 65° – 70°C. This temperature was held by cooling since the reaction is exothermic. 25 g (0.1441 mol) of 2-methyl-3- methoxy-4-chloropyridine N-oxide (X) were added over a period of about 70 minutes. Once the addition was completed, the reaction was held at 65° – 70°C for a further 2h 20 minutes and after this time it was allowed to cool down to below 65°C and 90 ml of methanol were added gradually, while holding the temperature below 65°C. The resulting reaction mass was distilled at reduced pressure in a rotavap to remove the volatile components and the residue containing compound (IX) was used as such for the following reaction. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), showed a single spot at Rf – 0.82, indicating that the reaction has been completed.
Example 2. – Preparation of compound fVIII

(IX) (VIII)
11.5 ml methanol and 11.5 ml of water were added over the crude residue from Example 1 containing compound (IX), and thereafter, while holding the temperature to between 25° and 30°C with a water bath, the residual acetic acid contained in the crude residue was neutralized by the addition of 33% aqueous NaOH. Once the residual acid had been neutralized, 19 ml (0.2136 mol) of the 33% aqueous NaOH were added over 20 minutes, while holding the temperature to between 25° and 30°C, and, on completion of the addition, the hydrolysis reaction at pH 11.7 – 11.8 was held for 2h 30 minutes, to between 25° and 30°C. On completion of the reaction, the pH was adjusted to 7.0 – 7.5 by the addition of HC1 35%, while holding the temperature to 25°C. Thereafter, 50 ml of methylene chloride were added and, after stirring and allowing to rest, the phases were decanted. A further five extractions were carried out with 30 ml methylene chloride each and the pooled organic phases were dried with anhydrous sodium sulfate, were filtered and washed, and were evaporated at reduced pressure in a rotavap, providing a solid residue having a melting point around 73°C and containing compound (VIII). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.55, showing that the reaction was complete. The thus obtained crude residue was used as such in the following reaction.
Example 3. – Preparation of compound (VI)

24.5 g of the residue obtained in Example 2, containing approximately 0.142 mol of the compound 2-hydroxymethyl-3-methoxy-4-chloropyridine (VIII), were mixed with 0.5 ml of DMF and 300 ml of anhydrous methylene chloride, to give a brown solution which was cooled to 0° – 5°C in an ice water bath. Thereafter, a solution of 11.5 ml (0.1585 mol) of thionyl chloride in 50 ml of anhydrous methylene chloride was added over 20 minutes, while holding the above-mentioned temperature,. Once the addition was complete, the reaction was held at 0° – 5°C for a further 90 minutes and then 120 ml of water and NaOH 33% were added to pH 5 – 6, requiring approximately 29 ml of NaOH. The phases were then decanted and separated. The organic phase was extracted with a further 120 ml of water and the pooled aqueous phases were extracted with a further 4×25 ml of methylene chloride, in order to recover the greatest possible amount of product. The pooled organic phases were dried over anhydrous sodium sulfate, filtered and washed, and evaporated at reduced pressure in a rotavap, to give a residue containing the compound 2-chloromethyl-3- methoxy-4-chloropyridine (VI). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15:1), showed a main spot at Rf = 0.83, indicating that the reaction was complete. The thus obtained crude residue was used as such in the following reaction. Example 4. – Preparation of compound (III)
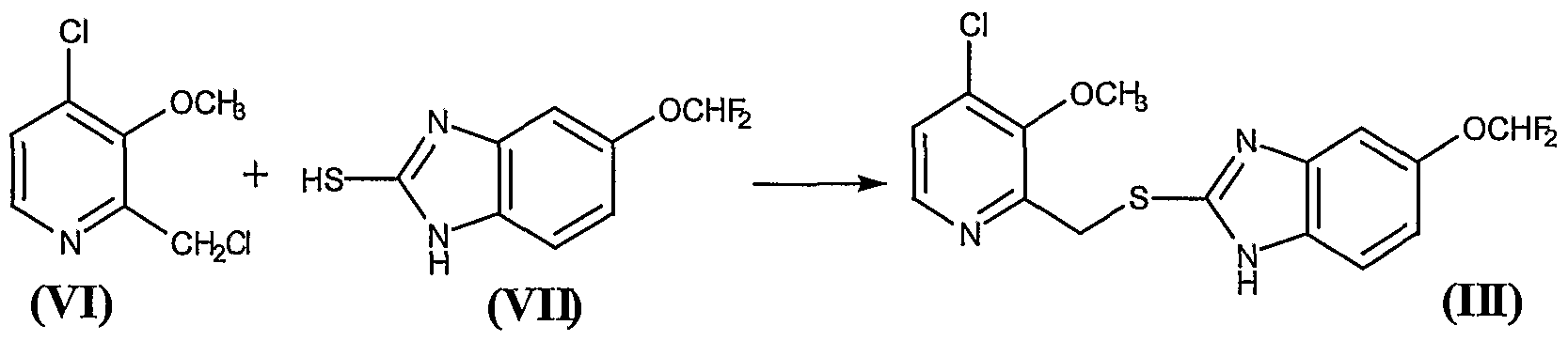
26.11 g of the residue obtained in the Example 3 containing approximately 0.136 mol of the compound 2-chloromethyl-3-methoxy-4- chloropyridine (VI) were mixed with 370 ml of methylene chloride, to give a brown solution over which were added, at 20° – 25°C, 29.3 g (0.136 mol) of 5-difluoromethoxy-2-mercaptobenzimidazole (VII) and 17.10 ml (0.136 mol) of tetramethylguanidine (TMGH). The mixture was stirred at this temperature for 2 hours, after which 450 ml of water were added, with the pH being held to between 9.5 and 10. Thereafter the phases were decanted and the organic phase was washed 5×50 ml of a IN NaOH aqueous solution and, thereafter, with 2×50 ml of water. The organic phase was treated with 50 ml of water and an amount of HC1 30% sufficient to adjust the pH to between 5 and 6. Thereafter, the phases were decanted, and the organic phase was dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a solid residue of melting point 64° – 73 °C that contains the compound (III). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), presented a main spot at Rf = 0.52. Yield 82%. The thus obtained compound 5-(difluoromethoxy)-2-[[(3-methoxy-4-chlorine-2 pyridinyl)methyl]mercapto]- lH-benzimidazole (III) was used as such in the following reaction Example 5. – Preparation of compound (IV)

25.8 g (0.0694 mol) of the compound (III) obtained in the Example 4 were mixed with 88 ml of methanol, to give a brown solution to which 3.7 ml of water, 0.99 g of ammonium molybdate and 0.78 g of sodium carbonate were added. The system was cooled to 0°C – 5°C, 3.4 ml (0.0756 mol) of 60% hydrogen peroxide were added, and the reaction mixture was held at 0°C – 5°C for 1 – 2 days, the end point of the reaction being checked by thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: l).
During the reaction the presence of hydrogen peroxide in the reaction medium was controlled by testing with potassium iodide, water and starch. When effected on a sample containing hydrogen peroxide, it provides a brown-black colour. If the assay is negative before the chromatographic control indicates completion of the reaction, more hydrogen peroxide is added.
On completion of the reaction, 260 ml of water were added, the system was cooled to 0°C – 5°C again and the mixture was stirred for 2 hours at this temperature. The solid precipitate was filtered, washed with abundant water, and dried at a temperature below 60°C, to give 5-(difluoromethoxy)-2-[[(3- methoxy-4-chlorine-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole (IV), melting point 130° – 136°C, with an 83.5% yield. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.5.
Compound (IV) can be purified, if desired, by the following crystallization method:
5 g of crude product was suspended in 16 ml of acetone and was heated to boiling until a dark brown solution was obtained. Thereafter the thus obtained solution was allowed to cool down to room temperature and then was then chilled again to -20°C, at which temperature the mixture was held for 23 hours without stirring. Thereafter the solid was filtered and washed with 6×4 ml of acetone chilled to -20°C. Once dry, the resulting white solid weighed 2.73 g, had a point of melting of 142°C and gave a single spot in thin layer chromatography. The IR spectrum of the compound on KBr is given in Figure 1.
The acetonic solution comprising the mother liquors of filtration and the washes was concentrated to a volume of 20 ml and a further 5 g of crude compound were added. The above described crystallization process was repeated to obtain a further 4.11 g of purified product of characteristics similar to the previous one.
The acetonic solution from the previous crystallization was concentrated to a volume of 17 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 2.91 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 15 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.3 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 16 ml and a further 4.36 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.62 g of purified product of similar characteristics to the previous ones.
Finally, the acetonic solution from the previous crystallization was concentrated to a volume of 10 – 12 ml and held at -20°C for two days without stirring. Thereafter, the solid was filtered and washed with 5×3 ml of acetone chilled to -20°C. Once dry, the solid weighed 1.26 g and had similar characteristics to the previous ones.
The total yield of all the crystallizations was 80%.
Example 6. – Preparation of pantoprazole

12.95 g (0.0334 mol) of compound (IV) purified by crystallization of Example 5 were mixed with 38 ml of N,N-dimethylacetamide and thereafter 7.03 g (0.1003 mol) of potassium methoxide were added, while holding the temperature to between 20°C and 30°C, whereby a dark brown mixture was obtained. The system was held at approximately 25°C for about 23 hours, after which, once the reaction was complete, the pH was adjusted to 7 with the addition of 3.82 ml of acetic acid. The N,N-dimethylacetamide was removed at reduced pressure at an internal temperature of not more than 75°C. 65 ml of water and 50 ml of methylene chloride were added over the thus obtained residue, followed by decantation of the phases. Once the phases were decanted, the aqueous phase was extracted a with further 3×25 ml of methylene chloride, the organic phases were pooled and the resulting solution dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a crude residue over which 55 ml of water were added, to give a suspension (if the product does not solidify at this point the water is decanted and a further 55 ml of water are added to remove remains of N,N-dimethylacetamide that hinder the solidification of the product). The solid was filtered and, after drying, 11.61 g of crude pantoprazole of reddish brown colour were obtained (Yield 90%). The thus obtained crude product was decoloured by dissolving the crude product in 150 ml of methanol, whereby a dark brown solution was obtained. 7.5 g of active carbon were added, while maintaining stirring for 45 minutes at 25°C – 30°C, after which the carbon was filtered out and the filter was washed. The methanol was then removed in the rotavap at reduced pressure, a temperature below 40°C. 10.33 g of a solid residue were obtained and were mixed with 14.9 ml of methylethylketone, and the suspension was heated to 45°C for about 10 minutes, after which it was cooled, first to room temperature and then to -20°C. This temperature was held over night and thereafter the solid was filtered, washed with 6×5 ml of methylethylketone chilled to -20°C. Once dry, 7.75 g of a white solid, melting point 140°C – 141 °C, were obtained. Thin layer chromatography on silica gel F254, eluting with CHCl3/MeOH (15: 1), gave a single spot at Rf =
0.41 and a IR spectrum corresponding identically with that of pantoprazole.
The ketonic solution comprising the mother liquors of filtration and the washes, was concentrated to 9.7 ml, was heated to 40°C, was held at this temperature for about five minutes and was then cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 4×2 ml of methylethylketone chilled to -20°C. Once dry, 0.42 g of a white solid of similar characteristics to the previous one was obtained.
The ketone solution from the previous treatment was concentrated to 3.1 ml, was heated to 40°C, was held to this temperature for about five minutes and then was cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 5×3 ml of methylethylketone chilled to – 20°C. Once dry, 0.41 g of a white-beige solid of similar characteristics to the previous one was obtained. The total yield, including purifications, was 67%.
If a whiter solid is desired, one or several washes can be carried with isopropyl acetate as follows: 6.6 g of pantoprazole from the methylethylketone treatment were suspended in 50 ml of isopropyl acetate. The system (white suspension) was stirred for about 30 minutes at 25°C, was then cooled to 0°C – 5°C, was stirred for about 15 minutes at this temperature and the solid was then filtered, was washed with 3×15 ml of isopropyl acetate. Once dry, 6.26 g of a pure white solid were obtained.
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Pantozol | Nycomed |
| Rifun | – “- | |
| France | Eupantol | Altana |
| Inipomp | Sanofi-Aventis | |
| United Kingdom | Protium | ALTANA |
| Italy | Pantekta | Abbott |
| Pantopan | Pharmacia | |
| Pantork | Altana | |
| USA | Protonix | Wyeth |
| Ukraine | Kontrolok | Nycomed Oranienburg GmbH, Germany |
| Nolpaza | Krka | |
| Pultset | Nobel Ilach Sanayi ve Ticaret AS, Turkey | |
| Proksium | JSC “Lubnyfarm”, Ukraine | |
| various generic drugs | ||
Formulations
-
ampoule 40 mg;
-
Tablets 40 mg
UV – spectrum
 |
||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| Solvent designation schedule |
Methanol |
Water |
0.1 M HCl |
0.1M NaOH |
|---|---|---|---|---|
| The absorption maximum | 289 nm | 291nm | Observed decay |
295 nm |
 |
391 | 346 | – | 418 |
| ε | 16600 | 14700 | – | 17700 |
IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wavenumber (cm -1 ) |
NMR Spectrum
will be added
Links
- EP 134 400 (Byk Gulden Lomberg; appl. 1.5.1984; CH-prior. 3.5.1983).
- US 4,555,518 (Byk Gulden Lomberg; 26.11.1985; appl. 1.5.1984; CH-prior. 3.5.1983).
- US 4,758,579 (Byk Gulden Lomberg; 19.7.1988; appl. 28.4.1987; CH-prior. 16.6.1984).
-
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
-
NIST / EPA / NIH Mass Spectral Library 2008
-
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
-
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
References
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
- [Dr. John Cooke, chair of Methodist Hospital’s cardiovascular services] [Houston Chronicle Health Zone dated Thursday, July 11, 2013 chron.com/refluxmeds] (Journal: Circulation)
- Jump up^ Meyer, U A (1996). “Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs”. European journal of gastroenterology & hepatology8 (Suppl 1): S21–25. doi:10.1097/00042737-199610001-00005.
- Steinijans, V. W.; Huber, R.; Hartmann, M.; Zech, K.; Bliesath, H.; Wurst, W.; Radtke, H. W. (1996). “Lack of pantoprazole drug interactions in man: An updated review”. International Journal of Clinical Pharmacology and Therapeutics 34 (6): 243–262. PMID 8793611.
- Sachs G, Shin JM, Hunt R (December 2010). “Novel approaches to inhibition of gastric acid secretion”. Curr Gastroenterol Rep 12 (6): 437–47. doi:10.1007/s11894-010-0149-5.PMC 2974194. PMID 20924727.
- Teva Announces Launch Of Generic Protonix Tablets
- Jump up^ Rubenstein, Sarah (29 January 2008). “Wyeth Plans Generic Protonix; Litigation With Teva to Continue”. The Wall Street Journal. p. D9. Retrieved 25 October 2009.
- Jump up^ “Nycomed and Wyeth announce launch of an own generic version of PROTONIX – lawsuit to defend patent continues”. Retrieved 25 October 2009.[dead link]
- Jump up^ IntelliPharmaCeutics Press Release