













Mavatrep; UNII-F197218T99; Mavatrep (USAN); JNJ-39439335; 956274-94-5;
2-(2-(2-(2-(4-trifluoromethylphenyl)vinyl)-1H-benzimidazol-5-yl)phenyl)propan-2-ol
(E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
Phase I Musculoskeletal pain; Pain
- 01 Mar 2013 Janssen Research and Development completes a phase I trial in Japanese and Caucasian adult male volunteers in the US (NCT01631487)
- 01 Mar 2013 Janssen completes enrolment in its phase I trial for Pain (in volunteers) in the USA (NCT01631487)
- 05 Feb 2013 Janssen Research and Development initiates enrolment in a phase I trial for Pain (Japanese and Caucasian volunteers) in USA (NCT01631487)
- Originator Johnson & Johnson Pharmaceutical Research & Development
- Developer Janssen Research & Development
- Class Analgesics; Benzimidazoles; Small molecules
- Mechanism of Action TRPV1 receptor antagonists
| PHASE 1 Johnson & Johnson Pharmaceutical Research & Development, L.L.C. |
|
| Public title: | A Clinical Study to Investigate the Effect on Pain Relief of a Single Dose of JNJ-39439335 in Patients With Chronic Osteoarthritis Pain of the Knee |
http://clinicaltrials.gov/ct2/show/NCT01006304
http://apps.who.int/trialsearch/trial.aspx?trialid=NCT00933582
http://www.ama-assn.org/resources/doc/usan/mavatrep.pdf SEE STRUCTURE IN THIS FILE
MAVATREP IS JNJ-39439335
—
(E)-2-(2-(2-(4-(trifluoromethyl)styryl)-1H-benzo[d]imidazol-6-yl)phenyl)propan-2-ol hydrochloride
956282-89-6 CAS NO OF HCl SALT





CLICK ON IMAGE FOR CLEAR VIEW
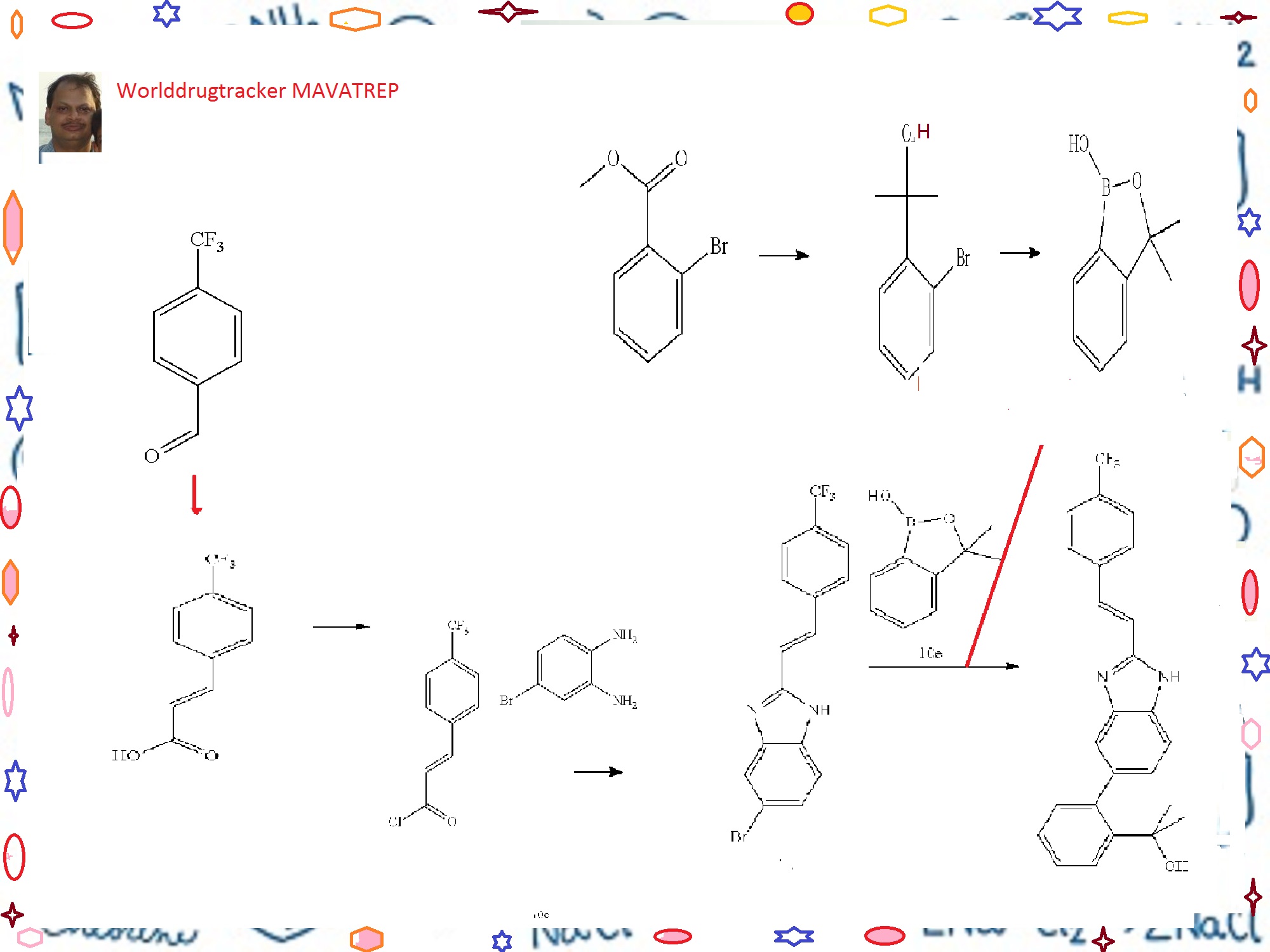
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0278]A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
-
[0000]
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0279]A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
-
[0280]To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
-
[0281]Hz), 7.56-7.62 (m, 3H), 7.50-7.52 (m, 1H), 7.34 (d, 1H, 16.7 Hz).
-
[0000]

Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0282]To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
-
[0000]
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0283]To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
-
[0000]
Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
[0284]To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.73 (m, 1H,), 7.90 (d, 2H, J=8.2 Hz), 7.85 (dd, 1H, J=8.0, 0.6 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.74 (d, 1H, J=16.8 Hz), 7.59-7.47 (m, 1H), 7.41 (s, 1H), 7.37-7.32 (m, 2H), 7.21 (dt, 1H, J=1.2, 7.4 Hz), 7.06 (s, 1H), 7.02 (d, 1H, J=7.4 Hz), 4.85 (s, 1H), 1.21 (s, 6H).
-
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
-
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0286]A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0287]To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
-
[0288]A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0289]A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0290]A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
-
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
-
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
//////////