












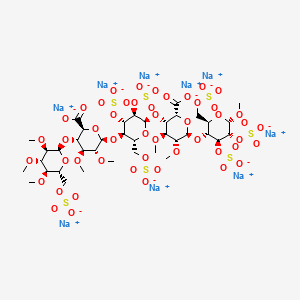
IDRAPARINUX
Nonasodium (2S,3S,4S,5R,6R)-6-[(2R,3R,4S,5R,6R)-6-[(2R,3S,4S,5R,6R)-2-carboxy-4,5-dimethoxy-6-[(2R,3R,4S,5R,6S)-6-methoxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxy-4,5-dimethoxy-3-[(2R,3R,4S,5R,6R)-3,4,5-trimethoxy-6-(sulfooxymethyl)oxan-2-yl]oxyoxane-2-carboxylic acid |
| CAS number | 149920-56-9 |
|---|
| Formula | C38H55Na9O49S7 |
|---|---|
| Mol. mass | 1727.17683 g/mol |
CAS 162610-17-5 (free acid)
SANORG34006, SR-34006, SanOrg 34006, SanOrg-34006, UNII-H84IXP29FN, AC1MJ0N4, Org-34006
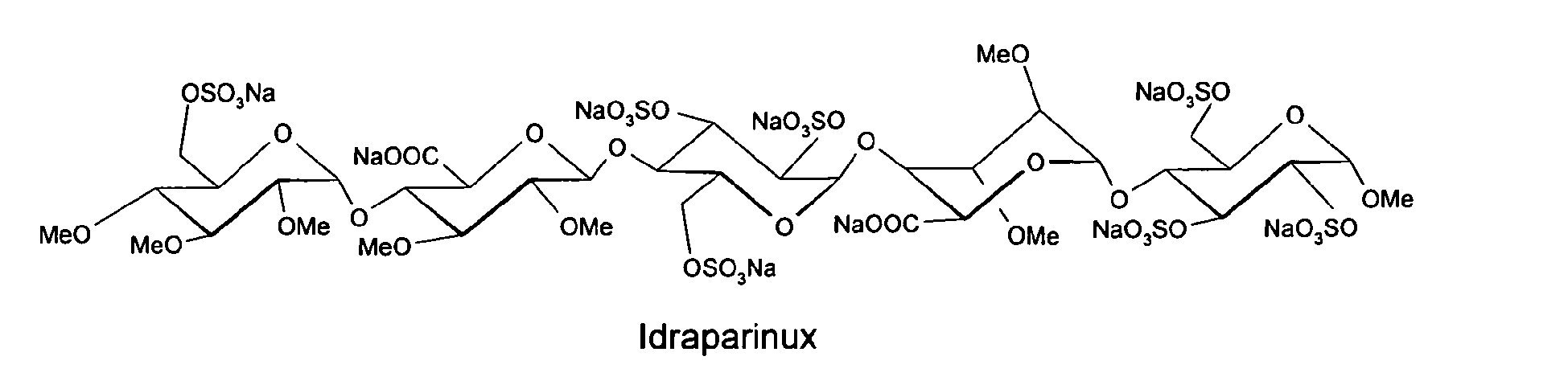
Methyl O-2,3,4-tri-O-methyl-6-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-beta-D-glucopyranuronosyl-(1–4)-O-2,3,6-tri-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-alpha-L-idopyranuronosyl-(1–4)-2,3,6-tri-O-sulfo-alpha-D-glucopyran
Sanofi-Syn(Originator), Organon (Codevelopment) , PHASE 3
, PHASE 3
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
Idraparinux sodium is an anticoagulant medication in development by Sanofi-Aventis.[1]
It has a similar chemical structure and the same method of action as fondaparinux, but with an elimination half-life about five to six times longer (an increase from fondaparinux’s 17 hours to approximately 80 hours), which means that the drug should only need to be injected once a week.
As of July 2007, it has completed the Phase III clinical trial AMADEUS.
Idraparinux selectively blocks coagulation factor Xa.[2]
See Heparin: Mechanism of anticoagulant action for a comparison of the mechanism of heparin, low-molecular-weight heparins, fondaparinux and idraparinux.
Idraparinux sodium is a synthetic pentasaccharide with indirect coagulation factor Xa inhibitor activity. The drug candidate had been in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the once-weekly long-term treatment and secondary prevention of venous thromboembolic events in patients with pulmonary embolism (PE) and deep vein thrombosis (DVT), as well as for the prevention of thromboembolic complications related to atrial fibrillation (AF).
However, no recent development has been reported for this research. The oligosaccharide is delivered by subcutaneous injection. Unlike other products, idraparinux is administered once weekly rather than daily, thereby increasing patient convenience.
Originally developed under a collaboration between sanofi-sventis and Akzo Nobel’s human healthcare business Organon, all rights to idraparinux were transferred to Sanofi in January 2004 in exchange for revenues based on future sales.
Several synthetic pentasaccharides have been developed, such as Idraparinux, where all hydroxyl groups are methylated or sulphated, as illustrated below:

Initially, the firm Organon developed a way of synthesis for the preparation of the “active pentasaccharide”. This synthesis, using the 3-0-benzyl-1 ,2-0-isopropylidene-a-D- glucofuranose as substrate (Van Boeckel et al., J. Carbohydr. Chem. 1985, 4, p.293-321 ), comprises more than 50 steps, and the inversion of configuration of the C5 carbon is carried out by the opening of an epoxide. After a step of protection followed by a bromination, the G unit is thus obtained. It is well known that the synthesis of said G unit is very tedious, due to the number of steps for obtaining such unit and the known tendency of L-idose derivatives to exist as furanoses. After being coupled to the H unit, successive steps of protection-deprotection then an oxidation reaction carried out on C6 carbon, lead to the GH disaccharide.
In the preparation of Idraparinux, the synthesis of the disaccharide GH is nearly similar to the above synthesis of early synthetic pentasaccharides. The major innovation lies in the obtaining of disaccharide EF by epimerization of disaccharide GH. The coupling of both disaccharides leads to the tetrasaccharide EFGH, which is further coupled to the D unit for obtaining said pentasaccharide. The preparation of the disaccharide EF from GH allows notably the decrease of the total number of the steps to approximatively 25 (Petitou, M.; Van Boeckel, C.A. Angew. Chem., Int.Ed. 2004, 43, p.31 18-3133).
Hence, all current syntheses of the “active pentasaccharide” comprise a large number of steps and more particularly involves the complex synthesis of key L-iduronic acid derivative (G unit). Indeed, the preparation of the G unit of the “active pentasaccharide” of heparin has always been a limiting step in the synthesis of antithrombotic heparin derivatives.
Thus, there is still a need for a new efficient process of preparation of L-iduronic acid derivative, which would not possess the drawbacks established above and would be compatible with industrial scales. Besides, there is a need for such process which would in addition lead to an improved process of preparation of the “active pentasaccharide” constituting the heparin derivatives.
- Idrabiotaparinux, developed by sanofi-aventis, is the biotinylated pentasaccharide corresponding to the structure depicted below. The pentasaccharide structure of idrabiotaparinux is the same as idraparinux, another antithrombotic agent developed by sanofi-aventis (see structure below). However in idrabiotaparinux, the presence of a biotin hook covalently linked to the first saccharidic unit enables the compound to be neutralized by avidin or streptavidin, as described in the international patent application WO 02/24754 .

- In the EQUINOX trial, which enrolled patients with DVT treated for 6 months with equimolar doses of either idrabiotaparinux or idraparinux, idrabiotaparinux, with the same anti-activated factor X pharmacological activity (hereafter “anti-Xa activity”) as idraparinux, was shown to have a similar efficacy, but, surprisingly, a better safety with less observed bleedings, in particular major bleedings.
- Therefore, the subject-matter of the invention is the use of idrabiotaparinux for the manufacture of a medicament useful for the treatment and secondary prevention of thrombotic pathologies, wherein the use of idrabiotaparinux involves a decrease in the incidence of bleedings during said treatment.
- In other words, the invention relates to the use of idrabiotaparinux as an antithrombotic treatment, wherein said use minimizes the risk of bleedings during the antithrombotic treatment. Indeed, idrabiotaparinux enables to increase the benefit-risk ratio during the antithrombotic treatment.
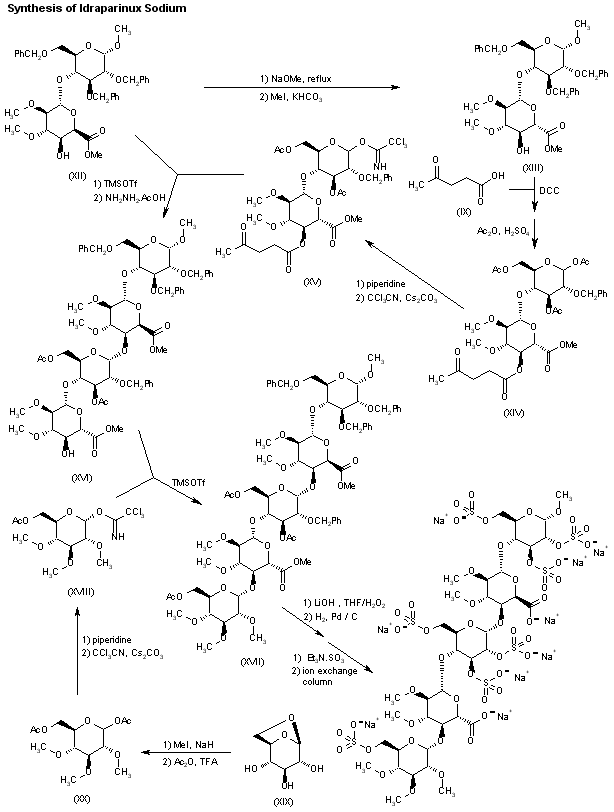
The L-ioduronic acid methyl ester derivative (XII) is then converted into its D-glucuronic acid methyl ester counterpart (XIII) by epimerization with NaOMe in refluxing MeOH, followed by esterification with MeI and KHCO3 in DMF.
Protection of the ester (XIII) with levulinic acid (IX) by means of DCC and DMAP in dioxane, followed by acetolysis of the anomeric center with sulfuric acid in acetic anhydride furnishes the disaccharide (XIV), which is then saponified with piperidine and subjected to reaction with trichloroacetonitrile and Cs2CO3 in THF to yield the imidate (XV).
Glycosylation of the disaccharide (XII) with the imidate (XV) by means of trimethylsilyl triflate in CH2Cl2, followed by removal of the levulinoyl group by means of hydrazine acetate, furnishes the tetrasaccharide (XVI), which is coupled with the glucosyl trichloroacetimidate (XVIII) by means of trimethylsilyl trifluoromethanesulfonate in CH2Cl2 providing the pentasaccharide (XVII).
Glucosyl imidate (XVIII) is prepared by methylation of 1,6-anhydroglucose (XIX) with MeI and NaH in DMF, followed by acetolysis with Ac2O/TFA to give compound (XX), which is treated with piperidine in THF and finally with trichloroacetonitrile in dichloromethane in the presence of Cs2CO3.
The pentasaccharide (XVII) is deprotected by saponification with LiOH in THF/H2O2, and then hydrogenated over Pd/C in tert-butanol/water to provide a fully deprotected pentamer, which is finally subjected to sulfation with triethylamine sulfur trioxide complex in DMF and converted into the corresponding sodium salt by elution in a Dowex 50 XW4-Na+ or a Mono-Q anion-exchange column.
……………..
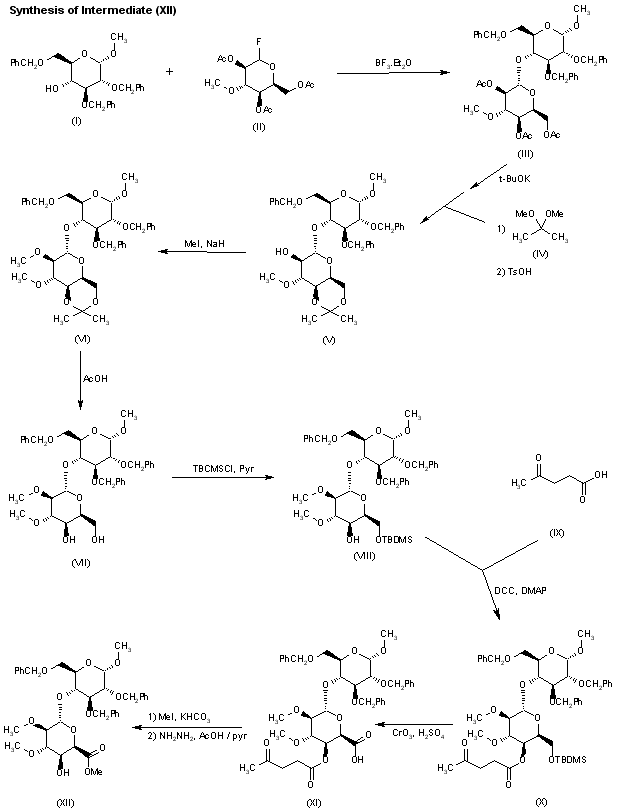
Glycosylation of sugar (I) with the idopyranosyl fluoride (II) by means of BF3.Et2O and molecular sieves in dichloromethane gives the disaccharide fragment (III), which is then converted into acetonide (V) by saponification of the ester functions with t-BuOK, followed by reaction with 2,2-dimethoxypropane (IV) in DMF and acidification with p-toluensulfonic acid. Methylation of acetonide (V) with MeI and NaH in DMF/MeOH provides the disaccharide (VI), which is then treated with HOAc to yield the 4′,6′-diol (VII). Selective silylation of the diol (VII) with tert-butyldimethylsilyl chloride (TBDMSCl) in pyridine leads to the 6′-O-TBDMS derivative (VIII), which is condensed with levulinic acid (IX) by means of dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) in dioxane to give the ester (X). Compound (X) is then submitted to simultaneous Jones oxidation and TBDMS removal with CrO3 and H2SO4/H2O in acetone to provide the iduronic acid derivative (XI), which is converted into the key intermediate (XII), first by esterification with MeI and KHCO3 in DMF and then by removal of the 4′-O-levulinoyl protecting group with HOAc and hydrazine hydrate in pyridine.
………………………
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
A crystalline form of the pentasaccharide methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose has now been isolated. This compound in its crystalline form has proven to be very useful for the preparation of idraparinux, since it makes it possible to obtain this product in a particularly interesting chemical yield and with a significant gain in quality, the purity being improved as regards the crude product obtained, as will be detailed hereinbelow. These gains in reaction yield and in purity for the production of idraparinux are considerable advantages from an industrial viewpoint, since improving the robustness of a process is a constant cause for concern, especially in the case of large-scale syntheses.
One subject of the invention is thus the compound methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic glucopyranose in crystalline form.
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I), corresponds to the following formula:

The compound of formula (I) in crystalline form according to the invention has a powder X-ray diffractogram whose characteristic lines are approximately at 12.009; 7.703; 7.300; 7.129; 5.838; 4.665; 4.476 and 3.785 angströms (interplanar distances). It also has a melting point of about 203° C. (203° C.±1° C.).
EXAMPLE 1 Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)

Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4 Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.

The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N′-dimethylformamide (6.6 mL) and then heated to 30° C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30° C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25° C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.
It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I″), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.
………………..
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
WAS PREPARED AS PER
- Example 3
methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt.
NOTE THIS IS ANALOGOUS PROCEDURE AND NOT SIMILAR
- Methyl O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranosyl-(1→4)-O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O-2,3,6-tri-O-acetyl-α-D-glucopyranoside (100 mg, 0.09 mmol), obtained by the known imidate coupling of the trichloroacetimidate of O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranoside and methyl O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O- 2,3,6-tri-O-acetyl-α-D-glucopyranoside, was dissolved in tetrahydrofuran (9 ml) and cooled to -5 °C. At this temperature a 30% aq. solution of hydrogen peroxide (4.5 ml) was added to the reaction mixture, and after 10 min a 1.25 M lithium hydroxide solution (4.7 ml) was added. The mixture was stirred for 1 h at -5 °C, after which time the temperature was raised to 0 °C and the mixture was stirred overnight. The reaction mixture was acidified with 6N hydrogen chloride at 0 °C to pH 1.5, after which the saponified compound was extracted with ethyl acetate. The organic layers were pooled, dried over magnesium sulfate, and evaporated to give 63 mg (84%) of methyl O-4-O-(4-nitrophenyl)-2,3-O-di-phenylmethy1-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside, which was dissolved in methanol (8 ml). 10% Pd on charcoal (63 mg) was added and the mixture hydrogenolyzed overnight. After filtration and evaporation 27 mg (50%) of methyl O-4-O-(4-aminophenyl)-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were obtained.
13 mg of methyl O-4-O-(4-aminophenyl)-O-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were dissolved in 2 ml of dry N,N-dimethylformamide, and under an atmosphere of nitrogen 148 mg of triethylamine sulfurtrioxide complex were added. The mixture was stirred overnight at 50 °C, after which an aq. solution of sodium hydrogen carbonate was added under ice cooling. The mixture was stirred for 1 h at room temperature, concentrated to a small volume and desalted on a Sephadex G-10 column with water. The crude product obtained was purified by HPLC using a Mono-Q anion exchange column to give 11 mg (37%) of methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +52.2° (c=0.67; water). Anomeric protons chemical shifts: 5.5; 5.17; and 5.15 ppm.
………………………………..
BMCL Volume 19, Issue 14, 15 July 2009, Pages 3875–3879
http://www.sciencedirect.com/science/article/pii/S0960894X0900482X


Final elaboration of the pentasaccharide 1. Reagents and conditions: (a) TMSOTf, Et2O, 4 Å MS, rt, 66% (28α), 15% (28β); (b) CAN, CH3CN, toluene, H2O, rt, 72%; (c) CCl3CN, DBU, CH2Cl2, rt, 98%; (d) TMSOTf, 4 Å MS, CH2Cl2, rt, 51% (73% based on recovery of 4); (e) Pd/C (10%), H2, t-BuOH, H2O, rt; (f) SO3·Et3N, DMF, 50 °C, 93% (2 steps).
The final elaboration of the pentasaccharide 1 was illustrated in IN ABOVE SCHEME Coupling of the glucopyranosyl trichloroacetimidate 6 with disaccharide acceptor 5 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in diethyl ether afforded the desired α-coupled trisaccharide 28α in a yield of 66%, together with 15% of the separable β-coupled product 28β. The anomeric 4-methoxyphenyl group in trisaccharide 28α was removed with CAN, and the resulting lactol was readily converted into the trisaccharide trichloroacetimidate 3. Coupling of donor 3 with the disaccharide acceptor 4 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in dichloromethane afforded the fully protected pentasaccharide 2 in 51% yield (73% based on recovery of 4). Finally, pentasaccharide 2 was subject to hydrogenolysis of the benzyl protecting groups. The highly polar product without purification was O-sulfated directly with triethylamine-sulfur trioxide complex to afford the sulfated pentasaccharide 1 in an excellent yield of 93% (for two steps).
Summarizing, the potent anti-thromboembolic pentasaccharide Idraparinux (1) was synthesized in total 51 steps and in 4% overall yield from d-glucose and methyl α-d-glucopyranoside.18 The synthetic route is convergent with a linear sequence of 27 steps, and the transformations are scalable. The 4-methoxyphenol glycoside intermediates are easy to be purified by crystallization.
Compound 1: ![]() 54.2 (c 1.0, H2O);
54.2 (c 1.0, H2O);
1H NMR (400 MHz, D2O) δ 3.27 (t, J = 8.4 Hz, 1H), 3.30–3.38 (m, 2H), 3.47 (s, 3H), 3.53 (s, 3H), 3.56 (s, 6H), 3.58 (s, 3H), 3.62 (s, 3H), 3.63 (s, 3H), 3.64 (s, 6H), 3.75 (d, J = 10.0 Hz, 1H), 3.83–3.97 (m, 4H), 3.98 (t, J = 8.8 Hz, 1H), 4.06–4.18 (m, 3H), 4.19–4.45 (m, 8H), 4.56 (br t, J = 9.6 Hz, 1H), 4.65 (t, J = 9.2 Hz, 1H), 4.66 (d, J = 7.6 Hz, 1H), 5.00 (br s, 1H), 5.11 (br s, 1H), 5.17 (d, J = 3.6 Hz, 1H), 5.43 (d,J = 3.2 Hz, 1H), 5.47 (d, J = 4.0 Hz, 1H);
ESI-MS m/z 774.1 [M−8Na+6H]2−, 763.0; [M−9Na+7H]2−, 508.5 [M−9Na+6H]3−.
………………….
The process of preparation of Idraparinux having the following formula:

may comprise the following steps :
1 ) preparation a compound of formula (IXB)

(IXB) wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, by the process according to the invention;
2) epimerisation of the disaccharide (IXB) so as to form disaccharide D of formula :

3) protection of the 4′-OH of D with a levulinoyl ester;
4) acetolysis of the disaccharide resulting from step 3), followed by preparation of the corresponding imidate;
5) coupling the disaccharide imidate resulting from step 4) with (IXB) obtainable by the process of the invention, wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, to obtain a tetrasaccharide;
6) coupling the fully protected tetrasaccharide with a monosaccharide glycosyl imidate;
7) deprotection of the protecting groups by the successive saponification and hydrogenolysis;
8) sulfation of the hydroxyl groups.
In one embodiment, the present invention concerns a process of preparation of Idraparinux:

said process comprising the following steps:
preparation of a compound of formula (VI) such as defined above, from a compound of formula (V) such as defined above; preparation of a compound of formula (VII) such as defined above, from a compound of formula (VI) such as defined above;
preparation of a compound of formula (VIII) such as defined above, from a compound of formula (VII) such as defined above;
– preparation of a compound of formula (IX) such as defined above, from a compound of formula (VIII) such as defined above;
wherein in compounds of formulae (V), (VI), (VII), (VIII) and (IX), R-i , R2, R3 and X are as defined above, Ra is methyl, Rb is methyl, Rc is methyl, Rd is methyl and R’ is the monosaccharide of formula :

wherein T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl. The inventors advantageously found that the process of preparation of Idraparinux comprising the decarboxylation/intramolecular cyclisation tandem reaction, which allows the inversion of configuration of C5 carbon of the compound of formula (VI), is more efficient than the processes previously described in the literature. Indeed, the process according to the invention allows advantageously a significant decrease of the number of steps and thus an improvement of the overall yield. Thus, the process of preparation of Idraparinux may be carried out in industrial scales. The inventors found an efficient process of preparation of Idraparinux.
According to another object, the present invention concerns the use of compounds of formulae (V), (VI), (VI I), (VIII) and (IX), as intermediates for the preparation of Idraparinux. In particular, the present invention concerns the use of a compounds of formulae (VB), (VI B), (VI IB), (VI I IB) and (IXB), as intermediates for the preparation of Idraparinux The invention is further illustrated but not restricted by the description in the following examples. Example 1 :
Preparation of Methyl-4,6-0-benzylidene-a-D-glucopyranoside (la)
CHC13)

Tf = 166-167°C (litt. 165-166°C) To a solution of benzaldehyde (400 mL, 3.94 mol, 5.9 eq.) was added zinc chloride (100.3 g, 0.74 mol, 1 .1 eq.) under vigorous stirring. After homogenization of the solution methyl- a-D-glucopyranoside (129.6 g, 0.67 mol, 1.0 eq.) was added portionwise. After 16 hours stirring at room temperature the reaction mixture was diluted with diethyl ether (100 mL). The mixture was then poured dropwise and under vigorous stirring in a solution containing ice water (1 .5 L) and hexane (350 mL). The precipitate was filtered, washed with diethyl ether (3 x 300 mL) and dried under vacuum over KOH. The product was then recrystallised from CH2CI2 (720 mL) and washed with a Et20/CH2CI2 solution (75:25, 2 x 200 mL). The filtrate was repeatedly recrystallised five times from CH2CI2 to afford compound la as white crystals (136.97 g, 0.49 mol, 72%).
1H NMR (CDCI3, 250 MHz): δ 2.35 (d, JCH-OH = 9.2 Hz, 1 H, OH), 2.83 (d, JCH-OH = 2.2 Hz, 1 H, OH), 3.46 (s, 3H, -OCH3), 3.43-3.46 (m, 1 H, H-4), 3.63 (td, JCH–OH = ^2,3 = 9.2 Hz, J1 2 = 3.9 Hz, 1 H, H-2), 3.70-3.81 (m, 2H, H-5, H-6), 3.93 (td, J = 9.2 Hz, JCH–OH = 2.2 Hz, 1 H, H- 3), 4.29 (m, 1 H, H-6 ), 4.79 (d, J1i2 = 3.9 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.35-7.38 (m, 3H, HAr), 7.47-7.51 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.6 (-OCH3), 62.5 (C-5), 69.0 (C-6), 71.1 (C-3), 72.9 (C- 2), 81 .0 (C-4), 99.9 (C-1 ), 102.0 (Ph-CH), 126.4, 128.5, 129.4, 137.1 (6xCAr). IR (film) v (cm“1): 3369 (O-H). Preparation of Methyl-2,3-di-0-methyl-4,6-0-benzylidene-a-D-glucopyranoside (Ma)
13)

°C)
To a solution of compound la (47.60 g, 0.17 mol, 1.0 eq.) in anhydrous THF (750 mL) under an argon atmosphere and cooled to 0°C, was added portionwise sodium hydride (60%, 16.93 g, 0.42 mol, 2.5 eq.). After 20 minutes methyl iodide was added dropwise (30 mL, 0.48 mol, 2.8 eq.) and the reaction mixture was allowed to reach room temperature. After 16 hours, methanol was added portionwise (75 mL) and the solution was stirred for another 15 minutes before being concentrated. The resulting residue was dissolved in EtOAc (400 mL) and washed with water (2 x 250 mL). The organic layer was dried (MgS04), filtered and concentrated. The resulting solid was dissolved in diethyl ether (1000 mL), hexane was added (400 mL) and the solvent was partially evaporated at low temperature. The crystals obtained were washed with hexane and the filtrate once again partially evaporated, filtered and the precipitate washed with hexane. The combined precipitates afforded compound Ma as white crystals (46.10 g, 0.15 mol, 88%).
1H NMR (CDCI3, 250 MHz): δ 3.30 (dd, J2-3 = 9.1 Hz, J1-2 = 3.7 Hz, 1 H, H-2), 3.44 (s, 3H, – OCH3), 3.49-3.87 (m, 4H, H-4, H-5, H-6, H-3), 3.55 (s, 3H, -OCH3), 3.64 (s, 3H, -OCH3), 4.28 (dd, J6-6. = 9.1 Hz, J5-6 = 3.7 Hz, 1 H, H-6 ), 4.85 (d, J1-2 = 3.7 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.36-7.41 (m, 3H, HAr), 7.48-7.52 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.4, 59.5, 61.1 (3x-OCH3), 62.3 (C-5), 69.1 (C-6), 79.9 (C-3), 81 .5 (C-4), 82.2 (C-2), 98.5 (C-1 ), 101.4 (Ph-CH), 126.2, 128.3, 129.0, 137.4 (6xCAr).
Preparation of Methyl-2,3-di-0-methyl-a-D-glucopyranoside (Ilia)
13)

To a suspension of compound Ma (10.33 g, 33.29 mmol, 1.0 eq.) in methanol (150 mL) was added para-toluenesulfonic acid monohydrate (322 mg, 1 .69 mmol, 0.05 eq.). After 4 hours stirring at room temperature, sodium carbonate (300 mg) was added and the reaction mixture was stirred an additional 15 minutes before filtration through a pad of Celite®. Then the filtrate was concentrated and the residue obtained was dissolved in a mixture of distilled water/diethyl ether (3:1 , 150 mL). The organic layer was extracted with water (2 x 50 mL) then the combined aqueous phases were concentrated and dried one night under vacuum over KOH. The resulting residue was recrystallised from toluene using petroleum ether as a co-solvent. The crystals obtained were washed with hexane and dried under vacuum over KOH to obtain compound Ilia as white crystals (6.63 g, 29.83 mmol, 90%).
1H NMR (DMSO, 400 MHz): δ 3.03 (dd, J2-3 = 9.3 Hz, J1-2 = 3.5 Hz, 1 H, H-2), 3.12-3.20 (m, 2H, H-3, H-4), 3.27 (s, 3H, -OCH3), 3.32 (s, 3H, -OCH3), 3.30-3.33 (m, 1 H, H-5), 3.44 (s, 3H, -OCH3), 3.40-3.46 (m, 1 H, H-6), 3.62 (ddd, J6-6‘ = 1 1.6 Hz, JCH-OH = 5.7 Hz, J5-6 = 1 ,9 Hz, 1 H, H-6′), 4.52 (t, J = 5.7 Hz, 1 H, OH), 4.78 (d, Ji-2 = 3.5 Hz, 1 H, H-1 ), 5.09 (d,
13C NMR (DMSO, 100.6 MHz): δ 54.1 , 57.4, 60.0 (3x-OCH3), 60.6 (C-6), 69.5 (C-3), 72.5 (C-5), 80.9 (C-2), 82.8 (C-4), 96.4 (C-1 ).
IR (film) v (cm“1): 3419 (O-H).
Preparation of Methyl methyl-2,3-di-0-methyl-a-D-glucopyranosiduronate (IVa)
C13)

To a solution of compound Ilia (500 mg, 2.25 mmol, 1.0 eq.) in distilled water (15 mL) were successively added NaBr (50 mg, 0.49 mmol, 0.2 eq.) and TEMPO (7 mg, 0.05 mmol, 0.02 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 5.2 mL, 9.1 mmol, 4.0 eq.) was added. After 5 hours stirring at 0°C ethanol was added (96% v/v, 8 mL), then the pH was reduced to 2-3 by addition of HCI (10 % v/v). The solvent was evaporated and the residue obtained was suspended in methanol, filtered in order to remove the remaining salts and washed several times with dichloromethane and methanol. The filtrate was concentrated then dissolved, under an argon atmosphere, in dry methanol (40 mL). para-toluenesulfonic acid (85 mg, 0.45 mmol, 0.2 eq.) was added then the reaction mixture was heated under reflux overnight. The solvent was evaporated and the residue obtained was dissolved in EtOAc (60 mL). The organic layer was washed with a 5% aqueous NaHC03 solution (2 χ 20 mL) and with brine (1 χ 20 mL). The aqueous phase was extracted with dichloromethane (3 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. Column chromatography (hexane/ethyl acetate 50:50) gave compound IVa as a colourless oil (503 mg, 2.00 mmol, 89%).
1H NMR (CDCIs, 400 MHz): δ 3.10 (d, JCH-OH = 3.0 Hz, 1 H, OH), 3.26 (dd, J2-3 = 9.3 Hz, J1 -2 = 3.4 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.49-3.52 (m, 1 H, H-3), 3.50 (s, 3H, -OCH3), 3.62 (s, 3H, -OCH3), 3.74 (td, J = 9.5 Hz, JCH-OH = 3.0 Hz, 1 H, H-4), 3.82 (s, 3H, -OCH3), 4.14 (d, J4.5 = 9.6 Hz, 1 H, H-5), 4.91 (d, Ji-2 = 3.4 Hz, 1 H, H-1 ).
13C NMR (CDCI3, 100.6 MHz): δ 52.9, 56.0, 59.1 , 61 .3 (4x-OCH3), 70.6 (C-5), 71.7 (C-4), 80.9 (C-2), 81.8 (C-3), 98.1 (C-1 ), 170.9 (C=0).
IR (film) v (cm“1): 3475 (O-H), 1750 (C=0).
Preparation of Methyl methyl^-O-il’-ethoxy^’-propyn-l’-ylJ^.S-di-O-methyl-a-D- glucopyranosiduronate (Va)

To a solution of compound IVa (4.56 g, 18.21 mmol, 1.0 eq.) in chloroform (stabilised with amylene, 200 mL) were added, under an argon atmosphere, P205 (5.31 g, 36.29 mmol, 2.0 eq.) and propargylaldehyde diethylacetal (5.2 mL, 36.27 mmol, 2.0 eq.), then the reaction mixture was heated at 60°C. After 4 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (300 mL), washed with a 5% NaHC03 aqueous solution (1 x 30 mL) and brine (1 x 30 ml_). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 20:80) afforded compound Va as a colourless oil (4.07 g, 12.24 mmol, 67%) in a diastereomeric mixture (64:36) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH), along with some unreacted compound IVa (1.17 g, 4.68 mmol, 26%).
1H NMR (CDCI3, 400 MHz): δ 1.18-1.25 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.56 (m, 1 H, H-C≡C-) (mixture), 3.26-3.31 (m, 1 H, H-2) (mixture), 3.43 (s, 3H, -OCH3) (major), 3.44 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, -OCH3) (mixture), 3.59 (s, 3H, -OCH3) (minor), 3.62 (s, 3H, -OCH3) (major), 3.47-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.65-3.73 (m, 1 H, -OCHaHbCH3) (mixture), 3.78 (s, 3H, -OCH3) (major), 3.80 (s, 3H, -OCH3) (minor), 3.78-3.86 (m, 1 H, H-4) (mixture), 4.15 (d, J4-5 = 10.0 Hz, 1 H, H-5) (major), 4.18 (d, J4-5 = 10.0 Hz, 1 H, H-5) (minor), 4.86-4.88 (m, 1 H, H-1 ) (mixture), 5.35 (d, J = 1.7 Hz, 1 H, EtO- CH) (minor), 5.58 (d, J = 1.7 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.0 (-OCH2CH3) (mixture), 52.6 (-OCH3) (major), 52.7 (- OCH3) (minor), 55.8 (-OCH3) (mixture), 59.2 (-OCH3) (major), 59.3 (-OCH3) (minor), 60.4 (-OCH2CH3) (major), 61 .3 (-OCH2CH3) (minor), 61.4 (-OCH3) (mixture), 70.1 (C-5) (minor), 70.2 (C-5) (major), 74.0 (H-C≡C-) (major), 74.2 (H-C≡C-) (minor), 76.4 (C-4) (minor), 76.7 (C-4) (major), 78.6 (H-C≡C-) (minor), 78.9 (H-C≡C-) (major), 81 .5 (C-2) (major), 81 .8 (C-2) (minor), 81.9 (C-3) (minor), 82.9 (C-3) (major), 92.6 (EtO-CH) (mixture), 97.9 (C-1 ) (minor), 98.0 (C-1 ) (major), 169.6 (C=0) (major), 169.9 (C=0) (minor). Elemental analysis: Calculated: C: 54.21 ; H: 7.28. Found: C: 54.17 ; H: 7.13.
ESI-MS (pos. mode): m/z = 355 [M+Na]+.
IR (film) v (cm“1): 1752 (C=0), 3266 (≡C-H). Preparation of 4-0-(1′-ethoxy-2′-propyn-1,-yl)-1 ,2,3-tri-0-methyl-a-D-gluco- pyranosiduronic acid (Via)

To a solution of compound Va (1.12 g, 3.37 mmol, 1.0 eq.) in EtOH/H20 (3:1 , 100 mL) was added sodium hydroxide (156 mg, 3.90 mmol, 1 .3 eq.). After 5 hours stirring at room temperature the solvent was evaporated. The residue obtained was dissolved in water (50 mL). The pH of the aqueous layer was reduced to 2-3 with a 5% citric acid aqueous solution, then the layer was saturated with sodium chloride before extraction with dichloromethane (10 x 20 mL). If necessary the pH was adjusted by addition of more citric acid aqueous solution. The combined organics were dried (MgS04), filtered and removed under vacuum. Compound Via was obtained without further purification as a colourless oil (1.020 g, 3.20 mmol, 95%), in a mixture of diastereomers (75:25) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH).
1H NMR (CDCIs, 400 MHz): δ 1.16-1.24 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.59 (d, J = 1 .6 Hz, 1 H, H-C≡C-) (major), 2.62 (s I, 1 H, H-C≡C-) (minor), 3.25-3.33 (m, 1 H, H- 2), 3.44 (s, 3H, -OCH3) (mixture), 3.51 (s, 3H, -OCH3) (mixture), 3.62 (s, 3H, -OCH3) (mixture), 3.54-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.68-3.77 (m, 1 H, -OCHaHbCH3) (mixture), 3.81-3.87 (m, 1 H, H-4) (mixture), 4.13-4.18 (m, 1 H, H-5) (mixture), 4.88-4.90 (m, 1 H, H-1 ), 5.45 (s I, 1 H, EtO-CH) (minor), 5.63 (d, J = 1.6 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 14.9 (-OCH2CH3) (mixture), 55.9 (-OCH3) (mixture), 59.2 (-OCH3) (minor), 59.3 (-OCH3) (major), 60.7 (-OCH2CH3) (mixture), 61 .2 (-OCH3) (minor), 61.3 (-OCH3) (major), 70.1 (C-5) (mixture), 74.3 (H-OC-) (major), 74.8 (H-OC-) (minor), 75.7 (C-4) (minor), 76.4 (C-4) (major), 78.5 (H-C≡C-) (minor), 78.8 (H-C≡C-) (major), 81.4 (C-2) (major), 81 .7 (C-2) (minor), 81 .8 (C-3) (minor), 82.9 (C-3) (major), 92.5 (EtO-CH) (mixture), 97.8 (C-1 ) (minor), 98.0 (C-1 ) (major), 173.8 (C=0) (major), 174.0 (C=0) (minor).
ESI-MS (pos. mode): m/z = 341 [M+Na]+. IR (film) v (cm“1): 1751 (C=0), 3268 (≡C-H).
Preparation of Methyl-4,7-anhydro-6-deoxy-6-methylene-7-ethoxy-2,3-di-0-methyl- a-L-/‘d -heptopyranoside (Vila)

To a solution of compound Via (1.89 g, 5.92 mmol, 1 .0 eq.) in anhydrous THF (40 mL) and cooled to 0°C, were added IBCF (0.84 mL, 6.48 mmol, 1.1 eq.) and N- methylmorpholine (0.72 mL, 6.55 mmol, 1.1 eq.). After 20 minutes stirring the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (1 .77 g, 1 1.80 mmol, 2.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 2 hours, anhydrous THF (80 mL) then ie f-butylthiol (0.28 mL, 2.61 mmol, 1.6 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 30 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13% v/v, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (2 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 25 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 75:25) afforded compound Vila as a colourless oil (218 mg, 0.79 mmol, 48%), in a mixture of diastereomers (67:33) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCIs, 400 MHz): δ 1.23 (t, J = 7.1 Hz, 3H, -OCH2CH3) (diastereomeric mixture), 3.13 (dd, J2-3 = 9.6 Hz, J1-2 = 3.0 Hz, 1 H, H-2) (minor), 3.30 (dd, J2-3 = 5.0 Hz, J1-2 = 1.6 Hz, 1 H, H-2) (major), 3.41 (s, 3H, -OCH3) (minor), 3.47 (s, 3H, -OCH3) (major), 3.50 (s, 3H, -OCH3) (mixture), 3.53 (s, 3H, -OCH3) (major), 3.55-3.61 (m, 1 H, -OCHaHbCH3) (mixture), 3.72 (dd, J2-3 = 5.0 Hz, J3-4 = 2.8 Hz, 1 H, H-3) (major), 3.78-3.90 (m, 1 H, – OCHaHbCH3) (mixture), 3.93-4.07 (m, 2H, H-3, H-4) (mixture), 4.59 (d I, J4-5 = 4.0 Hz, 1 H, H-5) (major), 4.62 (d, J1-2 = 1.7 Hz, 1 H, H-1 ) (major), (td, J4-5 = 7.9 Hz, J = 2.6 Hz, 1 H, H- 5), (minor), 4.79 (d, J1-2 = 3.0 Hz, 1 H, H-1 ) (minor), 5.35-5.57 (m, 3H, H-7, -C=CH2) (mixture). 13C NMR (CDCI3, 100.6 MHz): δ 15.3 (-OCH2CH3) (minor), 15.4 (-OCH2CH3) (major), 56.5 (-OCH3) (minor), 56.8 (-OCH3) (major), 58.6 (-OCH3) (major), 59.1 (-OCH3) (minor), 59.9 (- OCH3) (major), 60.3 (-OCH3) (minor), 63.3 (-OCH2CH3) (minor), 63.8 (-OCH2CH3) (major), 74.2 (C-5) (minor), 74.7 (C-5) (major), 76.4 (C-3) (major), 77.0 (C-4) (major), 77.7 (C-2) (major), 79.0 (C-3) (minor), 79.7 (C-4) (minor), 80.1 (C-2) (minor), 99.3 (C-1 ) (major), 99.5 (C-1 ) (minor), 102.2 (C-7) (major), 103.0 (C-7) (minor), 1 1 1.6 (-C=CH2) (minor), 1 15.2 (- C=CH2) (major), 147.4 (C-6) (minor), 148.1 (C-6) (major).
ESI-MS (pos. mode): m/z = 297 [M+Na]+.
Preparation of Methyl-4,7-anhydro-7-ethoxy-2,3-di-0-methyl-a-L-/ o-hepto- pyranosid-6-ulose (Villa)

Through a solution of compound Vila (449 mg, 1 .64 mmol, 1.0 eq.) in anhydrous dichloromethane (10 ml_), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (5 drops) was added and the solution was brought to room temperature. After 1 h15 the reaction mixture was concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 80:20) afforded compound Villa as a white solid (364 mg, 1.32 mmol, 80%), in a mixture of diastereomers (79:21 ) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCI3, 400 MHz): δ 1.24-1 .28 (m, 3H, -OCH2CH3) (diastereomeric mixture), 3.10 (dd, J2-3 = 10.2 Hz, J1-2 = 2.9 Hz, 1 H, H-2) (minor), 3.17 (dd, J2-3 = 9.4 Hz, J1-2 = 2.8 Hz, 1 H, H-2) (major), 3.38 (s, 3H, -OCH3) (major), 3.42 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, – OCH3) (mixture), 3.63 (s, 3H, -OCH3) (major), 3.66 (s, 3H, -OCH3) (minor), 3.48-3.73 (m, 3H, H-3 major, -OCHaHbCH3 major, -OCHaHbCH3 minor), 3.77-3.95 (m, 2H, -OCHaHbCH3 minor, -OCHaHbCH3 major), 4.07 (dd, J2-3 = 10.2 Hz, J3-4 = 7.7 Hz, 1 H, H-3) (minor), 4.34 (d, J4-5 = 9.1 Hz, 1 H, H-5) (minor), 4.39-4.44 (m, 2H, H-4 minor, H-5 major), 4.50 (dd, J3-4 = 9.5 Hz, J4-5 = 6.2 Hz, 1 H, H-4) (major), 4.76 (d, Ji-2 = 2.8 Hz, 1 H, H-1 ) (major), 4.79 (d, J1-2 = 2.9 Hz, 1 H, H-1 ) (minor), 4.89 (d, J = 1 ,1 Hz, 1 H, H-7) (minor), 4.93 (s I, 1 H, H-7) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.1 (-OCH2CH3) (minor), 15.2 (-OCH2CH3) (major), 56.7 (-OCH3) (minor), 57.2 (-OCH3) (major), 59.3 (-OCH3) (mixture), 59.8 (-OCH3) (major), 60.6 (-OCH3) (minor), 65.0 (-OCH2CH3) (minor), 65.5 (-OCH2CH3) (major), 70.2 (C-5) (major), 72.4 (C-5) (minor), 75.9 (C-4) (major), 79.2 (C-4) (minor), 79.4 (C-3) (major), 79.8 (C-2 major, C-3 minor), 80.2 (C-2) (minor), 96.1 (C-7) (major), 97.2 (C-7) (minor), 98.7 (C-1 ) (major), 99.0 (C-1 ) (minor), 205.3 (C-6) (minor), 205.6 (C-6) (major).
IR (film) v (cm“1): 1783 (C=0).
ESI-MS (pos. mode): m/z = 299 [M+Na]+, 331 [M+Na+MeOH]+.
Preparation of Methyl methyl-2,3-di-0-methyl-a-L-idopyranosiduronate (IXa)
; CHCI3)

To a solution of compound Villa (50 mg, 0.18 mmol, 1 .0 eq.) in dichloromethane (3 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 120 mg, 0.54 mmol, 3.0 eq.) and NaHC03 (20 mg, 0.23 mmol, 1 .3 eq.). After 3 hours stirring the solvent was removed under vacuum. The resulting residue was dissolved in EtOAc (30 mL), extracted with distilled water (2 x 10 mL), and the aqueous phase was concentrated. The crude mixture was dissoved in methanol (10 mL), para-toluenesulfonic acid monohydrate was added (4 mg, 0.02 mmol, 0.1 eq.) then the reaction mixture was heated to reflux and the reaction monitored by 1H NMR in deuterated methanol. After 8 hours the solvent was evaporated. The residue obtained was dissolved in DMF (5 mL) then triethylamine (28 μί, 0.20 mmol, 1 .1 eq.) and methyl iodide (56 μί, 0.90 mmol, 5 eq.) were added. After 3h30 the reaction mixture was concentrated, dissolved in EtOAc (30 mL) and the organic phase was washed with a 5% NaHC03 aqueous solution (2 x 10 mL), a 5% citric acid aqueous solution (2 x 10 mL) and brine (1 x 10 mL). The aqueous phase was extracted with dichloromethane (5 x 10 mL) and the combined organics were dried (MgS04), filtered and concentrated. Column chromatography (dichloromethane/ethyl acetate 85:15) afforded compound Xla as a colourless oil (25 mg, 0.10 mmol, 56%). 1H NMR (CDCI3, 400 MHz): δ 3.41 (d I, J2-3 = 3.5 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.56 (s, 3H, -OCH3), 3.57 (s, 3H, -OCH3), 3.69 (t, J2-3 = J3-4 = 3.5 Hz, 1 H, H-3), 3.75-3.78 (m, 1 H, OH), 3.80 (s, 3H, -OCH3), 3.97 (m, 1 H, H-4), 4.42 (d, J4-5 = 1 .6 Hz, 1 H, H-5), 4.61 (d,
13C NMR (CDCI3, 100.6 MHz): δ 52.4, 57.5, 58.4, 60.8 (4x-OCH3), 67.7 (C-4), 74.8 (C-5), 77.2 (C-2), 77.5 (C-3), 100.9 (C-1 ), 169.6 (C=0).
Elemental analysis: Calculated: C: 48.00 ; H: 7.25. Found: C: 47.62 ; H: 7.15.
ESI-MS (pos. mode): m/z = 272 [M+Na]+.
IR (film) v (cm“1): 3491 (O-H), 1765 (C=0). Example 2 :
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(2′,3′-di-0-methyl-p-D-glucopyranosyl- uronate)-a-D-glucopyranoside (IVb)

To a solution of the co

Me
(3.279 g, 5.00 mmol, 1.0 eq.) in a water/acetonitrile mixture (1 :1 , 300 mL) were added NaBr (105 mg, 1 .02 mmol, 0.2 eq.) and TEMPO (33 mg, 0.21 mmol, 0.04 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 1 1.5 mL, 20.08 mmol, 4.0 eq.) was added. After 3 hours stirring at 0°C, NaOCI was added anew (13% v/v, 1 1 .5 mL, 20.08 mmol, 4.0 eq.). After two more hours ethanol was added (96% v/v, 20 mL), then the pH was reduced to 2-3 by addition of HCI (10% v/v). The solvent was evaporated and the residue obtained was suspended in DMF (40 mL) then triethylamine (2.8 mL, 2.032 g, 20.0 mmol, 4.0 eq.) and methyl iodide (6.2 mL, 14.136 g, 99.6 mmol, 20.0 eq.) were added. After 4 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (1 χ 20 mL) and brine (1 χ 20 mL). The aqueous layer was extracted with dichloromethane (2 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. The residue obtained was dissolved in DMF (20 mL), then triethylamine (1.4 mL, 1 .016 g, 10.0 mmol, 2.0 eq.) and methyl iodide (3.1 mL, 7.068 g, 49.8 mmol, 10.0 eq) were added. After 60 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (2 x 20 mL) and brine (1 χ 20 mL). The organic layer was dried (MgS04), filtered and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 50:50) gave compound IVb as a colourless oil which was dissolved in a diethyl ether/hexane mixture and evaporated at room temperature to afford a white solid (2.500 g, 3.66 mmol, 73%).
1H NMR (CDCI3, 400 MHz): δ 2.92 (d, 1 H), 2.93-2.98 (m, 2H), 3.41 (s, 3H), 3.48-3.76 (m, 5H), 3.51 (s, 3H), 3.60 (s, 3H), 3.63 (s, 3H), 3.87-3.98 (m, 3H), 4.36-4.41 (m, 1 H), 4.49- 5.08 (m, 6H), 4.61 (d, 1 H), 7.24-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.09 ; H: 6.79. Found: C: 65.29 ; H: 6.96.
ESI-MS (pos. mode): m/z = 705 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2”-propyn-1 “-yl)-2′,3′- di-0-methyl-p-D-glucopyranosyluronate)-a-D-glucopyranoside (Vb)

E F To a solution of compound IVb (385 mg, 0.56 mmol, 1 .0 eq.) in chloroform (stabilised with amylene, 30 mL) were added, under an argon atmosphere, P205 (410 mg, 2.80 mmol, 5.0 eq.) and propargylaldehyde diethylacetal (0.4 mL, 2.79 mmol, 5.0 eq.), then the reaction mixture was heated at reflux. After 5 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (60 mL), washed with a 5% NaHC03 aqueous solution (1 x 15 mL) and brine (1 x 15 mL). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 70:30) afforded compound Vb as a colourless oil (275 mg, 0.36 mmol, 64%) in a diastereomeric mixture (64:36).
1H NMR (CDCI3, 250 MHz): δ 1.17-1 .27 (m, 3H), 2.55 (d, 0.36H), 2.57 (d, 0.64H), 2.92- 3.08 (m, 2H), 3.38 (s, 3H), 3.49 (s, 1.92H), 3.50 (s, 1.08H), 3.57 (s, 1.08H), 3.59 (s, 1.92H), 3.60 (s, 1.92H), 3.62 (s, 1.08H), 3.44-3.97 (m, 10H), 4.35 (t, 1 H), 4.46-4.76 (m, 6H), 5.03 (d, 1 H), 5.32 (d, 0.36H), 5.56 (d, 0.64H), 7.21-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.95 ; H: 6.85. Found: C: 65.92 ; H: 6.75.
ESI-MS (pos. mode): m/z = 787 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2″-propyn-1 ” 1 ‘,2′,3’-tri-0-methyl-a-D-glucopyranosiduronic acid)-a-D-glucopyranoside (Vlb)

E F
To a solution of compound Vb (1.02 g, 1.33 mmol, 1.0 eq.) in EtOH/H20 (1 :1 , 100 mL) was added sodium hydroxide (82 mg, 2.05 mmol, 1.5 eq.). After 3 hours stirring at room temperature sodium hydroxide was added anew (27 mg, 0.68 mmol, 0.5 eq.). After an additional hour stirring the solvent was evaporated. The residue obtained was dissolved in water (40 mL). The pH of the aqueous layer was reduced to 2-3 with a 10% HCI aqueous solution then the layer was saturated with sodium chloride before extraction with dichloromethane (3 x 20 mL). The combined organics were dried (MgS04), filtered and removed under vacuum. Compound VIb was obtained without further purification as a white solid (930 mg, 1.24 mmol, 93%), in a diastereomeric mixture (63:37).
1H NMR (CDCIs, 400 MHz): δ 1.17-1.28 (m, 3H), 2.65 (d, 0.63H), 2.68 (d, 0.37H), 2.90- 3.09 (m, 2H), 3.37 (s, 3H), 3.46 (s, 1.1 1 H), 3.57 (s, 1.89H), 3.58 (s, 1.1 1 H), 3.60 (s, 1.89H), 3.42-3.90 (m, 10H), 4.29 (d, 1 H), 4.46-4.97 (m, 7H), 5.44 (d, 0.37H), 5.61 (d, 0.63H), 7.28-7.44 (m, 15H).
ESI-MS (pos. mode): m/z = 773 [M+Na]+ , 795 [M-H+2Na]+ . ESI-MS (neg. mode): m/z = 749 [M-H]\
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-6′-deoxy-6′-methylene-7′- ethoxy-2 3′-di-0-methyl-α-L-/‘ o-heptopyranosyl)-α-D-glucopyranoside (Vllb)

To a solution of compound VIb (647 mg, 0.86 mmol, 1.0 eq.) in anhydrous THF (20 mL) and cooled to 0°C, were added IBCF (0.1 1 mL, 0.85 mmol, 1.0 eq.) and N- methylmorpholine (0.10 mL, 0.91 mmol, 1.1 eq.). After 10 minutes stirring, the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (512 mg, 3.43 mmol, 4.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 20 minutes anhydrous THF (100 mL) then ie f-butylthiol (0.18 mL, 1 .68 mmol, 2.0 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 15 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13%, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (1 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 15 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 90 :10 – 70:30) afforded compound Vllb as a colourless oil (251 mg, 0.36 mmol, 42%) in a mixture of diastereomers (61 :39).
1H NMR (CDCI3, 250 MHz): δ 1.18-1.29 (m, 3H), 2.90-3.07 (m, 1 H), 3.37 (s, 1.17H), 3.38 (s, 1 .83H), 3.46 (s, 3H), 3.54 (s, 1.83H), 3.60 (s, 1 .17H), 3.29-4.00 (m, 10H), 4.1 1 -4.26 (m, 1 H), 4.50-4.96 (m, 8H), 5.09-5.48 (m, 3H), 7.23-7.39 (m, 15H).
ESI-MS (pos. mode): m/z = 720 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-7′-ethoxy-2′,3′-di-0- methyl-a-L-/‘ o-heptopyranosid-6′-ulosyl)-a-D-glucopyranoside (Vlllb)

Through a solution of compound Vllb (145 mg, 0.21 mmol, 1 .0 eq.) in anhydrous dichloromethane (10 mL), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (4 drops) was added and the solution was brought to room temperature. After 30 min stirring the reaction mixture was concentrated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 60:40) afforded compound Vlllb as a white solid (100 mg, <67%) in a mixture of diastereomers (67:33).
1H NMR (CDCI3, 400 MHz): δ 1.21 -1 .28 (m, 3H), 2.93-3.07 (m, 1 H), 3.31-4.26 (m, 20H), 4.52-5.02 (m, 9H), 7.20-7.45 (m, 15H).
ESI-MS (pos. mode): m/z = 731 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(methyl 2′,3′-di-0-methyl-a-L- idopyranosiduronate)-a-D-glucopyranoside (IXb)

To a solution of compound Vlllb (62 mg, 87 μηηοΙ, 1 .0 eq.) in dichloromethane (5 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 58 mg, 259 μηηοΙ, 3.0 eq.) and NaHC03 (1 1 mg, 130 μηηοΙ, 1 .5 eq.). After 5 hours stirring the solvent was removed under vacuum. The reaction mixture was then dissolved in EtOAc (50 mL) and washed successively with a 5% NaHC03 aqueous solution (1 x 10 mL), a 5 % citric acid aqueous solution (1 x 10 mL) and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. The crude mixture was dissolved in anhydrous methanol (10 mL) and sodium methoxide was added to reach pH = 10. After 30 minutes stirring at room temperature the reaction mixture was neutralized with Dowex®, filtered through a pad of Celite®, and concentrated. The residue obtained was dissolved in DMF (10 mL) then triethylamine (13 μί, 93 μηηοΙ, 1.1 eq.) and methyl iodide (27 μί, 434 μηηοΙ, 5.0 eq.) were added. After 2h30 stirring the reaction mixture was concentrated, dissolved in EtOAc (40 mL) and washed with a 5% citric acid aqueous solution (2 x 10 mL), a 5% NaHC03 aqueous solution (2 x 10 mL), and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 60:40-50:50) afforded compound IXb as a colourless oil (12 mg, 18 μηηοΙ, 20% over two steps).
1H NMR (CDCIs, 400 MHz): δ 3.23 (s, 3H), 3.20-3.25 (m, 1 H), 3.36 (s, 3H), 3.43 (s, 3H), 3.46 (s, 3H), 3.33-3.58 (m, 3H), 3.60-3.69 (m, 2H), 3.72-3.95 (m, 4H), 4.53-4.60 (m, 4H), 4.68-4.97 (m, 4H), 5.14 (s, 1 H), 7.24-7.37 (15H).
ESI-MS (pos. mode): m/z = 705 [M+Na]+ .
……………
Volume 69, Issue 15, 15 April 2013, Pages 3149–3158
http://www.sciencedirect.com/science/article/pii/S0040402013003025
Abstract
Idraparinux, the fully O-sulfated, O-methylated, heparin-related pentasaccharide possessing selective factor Xa inhibitory activity, was prepared by two novel synthetic pathways. Each route was based on a 2+3 block synthesis utilizing the same l-iduronic acid-containing trisaccharide acceptor, which was glycosylated with either a glucuronide disaccharide donor or its non-oxidized precursor. The latter route, involving the oxidation of the glucose unit into d-glucuronic acid at a pentasaccharide level proved to be much more efficient, providing the target pentasaccharide in a reasonable overall yield.
Graphical abstract

- ……………………………
- SYNTHESIS
- US 20120041189 A1,
- http://www.patexia.com/us-publications/20120041189
- EXAMPLE 1Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4
Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.
The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N’-dimethylformamide (6.6 mL) and then heated to 30.degree. C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30.degree. C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25.degree. C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I”), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.
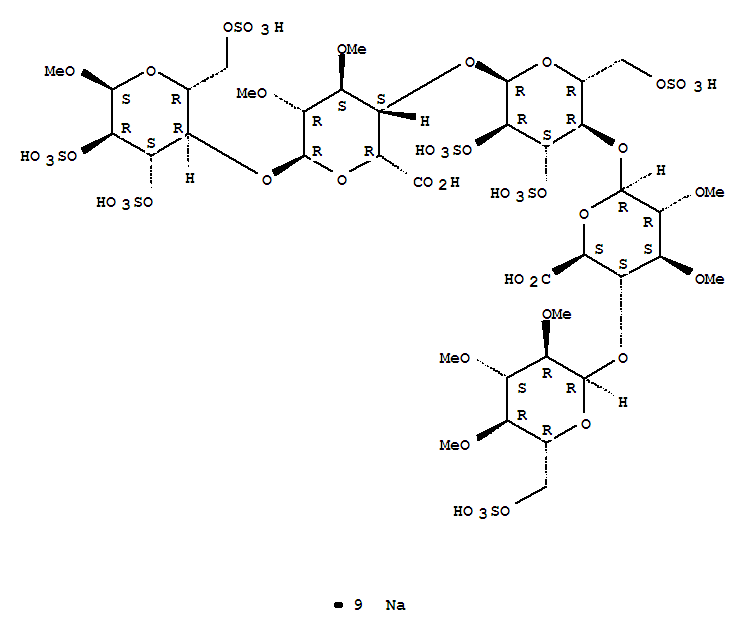
IDRAPARINUX
References
- Bousser MG, Bouthier J, Büller HR, et al. (January 2008). “Comparison of idraparinux with vitamin K antagonists for prevention of thromboembolism in patients with atrial fibrillation: a randomised, open-label, non-inferiority trial”. Lancet 371 (9609): 315–21. doi:10.1016/S0140-6736(08)60168-3.PMID 18294998.
- Buller HR, Cohen AT, Davidson B, et al. (September 2007). “Idraparinux versus standard therapy for venous thromboembolic disease”. N. Engl. J. Med. 357 (11): 1094–104. doi:10.1056/NEJMoa064247. PMID 17855670.
- Bioorg Med Chem1994,2,(11):1267
- Drugs Fut2002,27,(7):639
- EP 0454220, JP 1992225994, US 5378829, US 5382570, US 5529985, US 5773605
- EP 0529715
- Bioorg Med Chem Lett. 2009 Jul 15;19(14):3875-9. doi: 10.1016/j.bmcl.2009.03.155. Epub 2009 Apr 5.
- Chemistry – A European Journal, 2012 , vol. 18, 34 p. 10643 – 10652
- Magnetic Resonance in Chemistry, 2001 , vol. 39, 5 p. 288 – 293
- Tetrahedron, 2013 , vol. 69, 15 p. 3149 – 3158…….. MP 210-15 DEG CENT
-
- I. Capila, R.J. LinhardtAngew. Chem., Int. Ed., 41 (2002), p. 390
-
- L. RodenD.A. Lane, U. Lindahl (Eds.), Chemical and Biological Properties, Clinical Applications, CRC Press, Boca Raton, FL (1989), p. 1
-
- (a) C.A.A. van Boeckel, M. PetitouAngew. Chem., Int. Ed. Engl., 32 (1993), p. 1671
- (b) M. Petitou, C.A.A. van BoeckelAngew. Chem., Int. Ed., 43 (2004), p. 3118
-
- M. Petitou, B. Casu, U. LindahlBiochimie, 85 (2003), p. 83Article |
-
- (a) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.C. Jacquinet, P. Sinay, G. TorriCarbohydr. Res., 167 (1987), p. 67Article |
- (b) P.-A. Driguez, I. Lederman, J.-M. Strassel, J.-M. Herbert, M. PetitouJ. Org. Chem., 64 (1999), p. 9512
- (c) J. Choay, M. Petitou, J.C. Lormeau, P. Sinay, B. Casu, G. GattiBiochem. Biophys. Res. Commun., 116 (1983), p. 492Article |
- (d) P. Sinay, J.C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, G. TorriCarbohydr. Res., 132 (1984), p. C5
-
- (a) P. Westerduin, C.A.A. van Boeckel, J.E.M. Basten, M.A. Broekhoven, H. Lucas, A. Rood, H. van der Heijden, R.G.M. van Amsterdam, T.G. van Dinther, D.G. Meuleman, A. Visser, G.M.T. Vogel, J.B.L. Damm, G.T. Overklift
- Bioorg. Med. Chem., 2 (1994), p. 1267Article |
 PDF (1579 K)
PDF (1579 K)
- (b) J.M. Herbert, J.P. Herault, A. Bernat, R.G.M. van Amsterdam, J.C. Lormeau, M. Petitou, C. van Boeckel, P. Hoffmann, D.G. MeulemanBlood, 91 (1998), p. 4197
-
- (a) P. Prandoni, D. Tormene, M. Perlati, B. Brandolin, L. SpieziaExpert Opin. Investig. Drugs, 17 (2008), p. 773
- (b) A.S. Go, D.E. SingerLancet, 371 (2008), p. 278Article|
-
- I.M. Pinilla, M.B. Martinez, J.A. GalbisCarbohydr. Res., 338 (2003), p. 549Article |
-
- (a) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai, D.N. ReinhoudtChem. Eur. J., 5 (1999), p. 2722
- (b) J. Elhalabi, K.G. RiceCarbohydr. Res., 335 (2001), p. 159Article| PDF (177 K)
-
- S.D. Debenham, E.J. TooneTetrahedron: Asymmetry, 11 (2000), p. 385Article |
-
- A. Meijer, U. EllervikJ. Org. Chem., 69 (2004), p. 6249 and references therein
-
- P. Duchaussoy, G. Jaurand, P.-A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou, J.-M. HerbertCarbohydr. Res., 317 (1999), p. 63Article |
-
- J. Lee, X.-A. Lu, S.S. Kulkarni, Y. Wen, S.-C. HungJ. Am. Chem. Soc., 126 (2004), p. 476
-
- L.A.G.M. van den Broek, D.J. Vermaas, B.M. Heskamp, C.A.A. van BoeckelRecl. Trav. Chim. Pays-Bas., 112 (1993), p. 82
- R.R. Schmidt, J. MichelAngew. Chem., Int. Ed. Engl., 19 (1980), p. 731
-
- (a) F. Lin, W. Peng, W. Xu, X. Han, B. YuCarbohydr. Res., 339 (2004), p. 1219
- Article | PDF (270 K)
- (b) P.L. Anelli, C. Biffi, F. Montanari, S. QuiciJ. Org. Chem., 52 (1987), p. 2559
- (c) P.L. Anelli, S. Banfi, F. Montanari, S. QuiciJ. Org. Chem., 54 (1989), p. 2970
-
- (a) P. Bourhis, F. Machetto, P. Duchaussoy, J.-P. Herault, J.-M. Mallet, J.-M. Herbert, M. Petitou, P. Sinay
- For other examples of 4,6-locked idose glycosyl donors, see: Bioorg. Med. Chem. Lett., 7 (1997), p. 2843
- Article | PDF (244 K)
- (b) N. Barroca, J.-C. JacquinetCarbohydr. Res., 337 (2002), p. 673
- Article | PDF (300 K)
- (c) J. Kuszmann, G. Medgyes, S. BorosCarbohydr. Res., 339 (2004), p. 1569
- Article | PDF (352 K)
- (d) J. Tatai, P. FugediOrg. Lett., 9 (2007), p. 4647
| WO2002024754A1 | Sep 20, 2001 | Mar 28, 2002 | Akzo Nobel Nv | Polysaccharides with antithrombotic activity comprising at least a covalent bond with biotin or a biotin derivative |
| WO2006030104A1 * | Sep 7, 2005 | Mar 23, 2006 | Sanofi Aventis | Biotinylated hexadecasaccharides, preparation and use thereof |
| WO2007042469A2 * | Oct 6, 2006 | Apr 19, 2007 | Organon Nv | Anticoagulant antithrombotic dual inhibitors comprising a biotin label |
| EP0230023A2 * | Dec 19, 1986 | Jul 29, 1987 | Marion Merrell Dow Inc. | Pharmaceutical compositions for the enhancement of wound healing |
| EP0300099A1 * | Jul 20, 1987 | Jan 25, 1989 | Akzo N.V. | New pentasaccharides |
| EP0301618A2 * | Jul 4, 1988 | Feb 1, 1989 | Akzo N.V. | New pentasaccharides |
| EP0454220A1 * | Apr 16, 1991 | Oct 30, 1991 | Akzo Nobel N.V. | Carbohydrate derivatives comprising a trisaccharide unit |
| GB1110939A * | Title not available | |||
| US3017407 * | Aug 18, 1958 | Jan 16, 1962 | Riker Laboratories Inc | Process for producing polysulfuric acid esters of polysaccharides |
