














- 1.
Mather, R.; Karenchak, L. M.; Romanowski, E. G.; Kowalski, R. P. Am. J. Ophthalmol.2002, 133 ( 4) 463
- 2.
Corey, E. J.; Czakó, B.; Kürti, L. Molecules and Medicine; Wiley: NJ, 2007; p 135.
- 3.
Masuzawa, K.; Suzue, S.; Hirai, K.; Ishizaki, T. 8-Alkoxyquinolonecarboxylic acid and salts thereof excellent in the selective toxicity and process of preparing the same EP 0 230 295 A3, 1987.
- 4.
Niddam-Hildesheim, V.; Dolitzky, B.-Z.; Pilarsky, G.; Steribaum, G. Synthesis of Gatifloxacin WO 2004/069825 A1, 2004.
- 5.
Ruzic, M; Relic, M; Tomsic, Z; Mirtek, M. Process for the preparation of Gatifloxacin and regeneration of degradation products WO 2006/004561 A1, 2006.
- 6.
Iwata, M.; Kimura, T.; Fujiwara, Y.; Katsube, T. Quinoline-3-carboxylic acid derivatives, their preparation and use EP 0 241 206 A2, 1987.
- 7.
Sanchez, J. P.; Gogliotti, R. D.; Domagala, J. M.; Garcheck, S. J.; Huband, M. D.; Sesnie,J. A.; Cohen, M. A.; Shapiro, M. A. J. Med. Chem. 1995, 38, 4478
- 8.
Satyanarayana, C.; Ramanjaneyulu, G. S.; Kumar, I. V. S. Novel crystalline forms of Gatifloxacin WO 2005/009970 A1 2005.
- 9.
Takagi, N.; Fubasami, H.; Matsukobo, H.; (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture EP 0 464 823 A1, 1991.
………………………….
WO 2005009970
http://www.google.com/patents/WO2005009970A1?cl=en
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
STAGE – I:

Ethyl l-cyclopropyl-6,7-difluoro-8-met oxy l-Cycloproρyl-6, 7-difluoro-8-methoxy -4-oxo-l, -dihydro-3-quinoline -4-oxo-l, 4-dihydro-3-quinoline carboxylate carboxylic acid boron difluoride chelate
STAGE – II :

l-Cycloprop l-7- ( 3-methylpiperazin-l-yl.
6-fluoro~8-methoxy-4-oxo-l , 4-dihydro-3- carboxylicacid borondifluoride chelate quinoline carboxylicacid borondifluoride chelate
STAGE -III :

l-Cyclopropyl-7- (3- ethylpiperaz.in-l-yl . GATIFLOXACIN
-6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3- quinoline carboxylicacid borondifluoride chelate
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
Stage-2: Preparation of 1- Cyclopropyl-7- (3-methylpiperazin-l-yl) – 6-fluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylic acid boron difluoride chelate
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate.
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
……………………….
WO 2005047260
http://www.google.co.in/patents/WO2005047260A1?cl=en
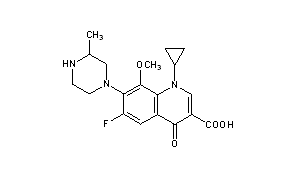
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:

European patent application EP-A-230295 discloses a process for obtaining gatifloxacin that consists on the reaction of compound (II) with 2-

In this process the gatifloxacin is isolated in the form of a hemihydrate after a laborious process of column chromatography and recrystallisation in methanol, which contributes towards making the final yield lower than 20% by weight. Moreover, in said process an undesired by-product is formed, resulting from demethylation at position 8 of the ring. European patent application EP-A-241206 discloses a process for preparing gatifloxacin, whose final steps are as follows:

(III) H ft N Me H DMSO
Gatifloxacin (I)

(IV) This process uses the intermediate compound (III) , which has been prepared and isolated in a separate operation, while the intermediate compound (IV) is also isolated before proceeding to its conversion into gatifloxacin by treatment with ethanol in the presence of triethylamine. The overall yield from these three steps is lower than 40%. These disadvantages — a synthesis involving several steps, low yields, and the need to isolate the intermediate products — hinder the production of gatifloxacin on an industrial scale. There is therefore a need to provide a process for preparing gatifloxacin with a good chemical yield, without the need to isolate the intermediate compounds and that substantially avoids demethylation in position 8 of the ring. The processes termed in English “one pot” are characterised in that the synthesis is carried out in the same reaction vessel, without isolating the intermediate compounds, and by means of successive addition of the reacting compounds. The authors of the present invention have discovered a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.

Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-cyclopropyl-6-fluoro- 8-methoxy-7-(3-methylpiperazin-1-yl)- 4-oxo-quinoline-3-carboxylic acid | |
| Clinical data | |
| Trade names | Zymar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605012 |
|
|
| Oral (discontinued), Intravenous(discontinued) ophthalmic |
|
| Pharmacokinetic data | |
| Protein binding | 20% |
| Half-life | 7 to 14 hours |
| Identifiers | |
| 112811-59-3 |
|
| J01MA16 S01AE06 | |
| PubChem | CID: 5379 |
| DrugBank | DB01044 |
| ChemSpider | 5186 |
| UNII | 81485Y3A9A |
| KEGG | D08011 |
| ChEBI | CHEBI:5280 |
| ChEMBL | CHEMBL31 |
| NIAID ChemDB | 044913 |
| Chemical data | |
| Formula | C19H22FN3O4 |
| 375.394 g/mol | |
PAPER

An improved process to obtain gatifloxacin (1) through use of boron chelate intermediates has been developed. The methodology involves an initial activation step which accelerates the formation of the first chelate under low-temperature conditions and prevents demethylation of the starting material. To increase the overall yield and to avoid the isolation and manipulation of the resulting intermediates, the process has been designed to be carried out in one pot. As a result, we present here an easy, scaleable and substantially impurity-free process to obtain gatifloxacin (1) in high yield.
A High-Throughput Impurity-Free Process for Gatifloxacin
DSC analysis showed two endothermic peaks at 166.2 °C (T onset = 164.3 °C) and 190.0 °C (T onset = 188.2 °C) and an exothermic one at 168.1 °C. The shape of this DSC curve is characteristic of a monotropic transition between crystalline forms
Although there are several hydrates described for gatifloxacin such as, among others, the hemimydrate, sesquihydrate, and pentahydrate(Raghavan, K. S.; Ranadive, S. A.;Gougoutas, J. Z.; Dimarco, J. D.; Parker, W. L.; Dovich, M.; Neuman, A.Gatifloxacin pentahydrate. WO 2002/22126 A1, 2002) , the Gatifloxacin obtained by the present procedure does not seem to form a stoichometric hydrate, but instead it retains moisture.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
Side-effects and removal from the market
A Canadian study published in the New England Journal of Medicine in March 2006 claims Tequin can have significant side effectsincluding dysglycemia.[2] An editorial by Dr. Jerry Gurwitz in the same issue called for the Food and Drug Administration (FDA) to consider giving Tequin a black box warning.[3] This editorial followed distribution of a letter dated February 15 by Bristol-Myers Squibb to health care providers indicating action taken with the FDA to strengthen warnings for the medication.[4] Subsequently it was reported on May 1, 2006 that Bristol-Myers Squibb would stop manufacture of Tequin, end sales of the drug after existing stockpiles were exhausted, and return all rights to Kyorin.[5]
Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
Contraindications
Availability
Gatifloxacin is currently available only in the US and Canada as an ophthalmic solution.
In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.

- The reaction of 1-bromo-2,4,5-trifluoro-3-methoxybenzene (I) with CuCN and N-methyl-2-pyrrolidone at 150 C gives 2,4,5-trifluoro-3-methoxybenzonitrile (II), which by treatment with concentrated H2SO4 yields the benzamide (III) The hydrolysis of (III) with H2SO4 -. water at 110 C affords 2,4,5-trifluoro-2-methoxybenzoic acid (IV), which by reaction with SOCl2 is converted into the acyl chloride (V). The condensation of (V) with diethyl malonate by means of magnesium ethoxide in toluene affords diethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) malonate (VI), which by treatment with p-toluenesulfonic acid in refluxing water gives ethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) acetate (VII). The condensation of (VII) with triethyl orthoformate in refluxing acetic anhydride yields 3-ethoxy -2- (2,4,5-trifluoro-3-methoxybenzoyl) acrylic acid ethyl ester (VIII), which is treated with cyclopropylamine (IX) to afford the corresponding cyclopropylamino derivative (X). The cyclization of (X) by means of NaF in refluxing DMF gives 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (XI), which is hydrolyzed with H2SO4 in acetic acid to yield the corresponding free acid (XII). Finally, this compound is condensed with 2-methylpiperazine (XIII) in hot DMSO.
 |
|
Title: Gatifloxacin
CAS Registry Number: 112811-59-3
CAS Name: 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid
Trademarks: Tequin (BMS); Zymar (Allergan)
Molecular Formula: C19H22FN3O4
Molecular Weight: 375.39
Percent Composition: C 60.79%, H 5.91%, F 5.06%, N 11.19%, O 17.05%
Literature References: Fluorinated quinolone antibacterial. Prepn: K. Masuzawa et al., EP 230295; eidem, US 4980470 (1987, 1990 both to Kyorin); J. P. Sanchez et al., J. Med. Chem. 38, 4478 (1995); of the sesquihydrate: T. Matsumoto et al., US5880283 (1999 to Kyorin). In vitro antibacterial activity: A. Bauernfeind, J. Antimicrob. Chemother. 40, 639 (1997); H. Fukuda et al., Antimicrob. Agents Chemother. 42, 1917 (1998). Clinical pharmacokinetics: M. Nakashima et al., ibid. 39, 2635 (1995). Clinical study in urinary tract infection: H. Nito, 10th Mediterranean Congr. Chemother. 1996, 327; in respiratory tract infection: S. Sethi, Expert Opin. Pharmacother. 4, 1847 (2003).
Properties: Pale yellow prisms from methanol as hemihydrate, mp 162°.
Melting point: mp 162°
Derivative Type: Sesquihydrate
CAS Registry Number: 180200-66-2
Manufacturers’ Codes: AM-1155
Molecular Formula: C19H22FN3O4.1½H2O
Molecular Weight: 384.40
Percent Composition: C 59.37%, H 6.03%, F 4.94%, N 10.93%, O 18.73%
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Quinolones and Analogs
|
References
- Burka JM, Bower KS, Vanroekel RC, Stutzman RD, Kuzmowych CP, Howard RS (July 2005). “The effect of fourth-generation fluoroquinolones gatifloxacin and moxifloxacin on epithelial healing following photorefractive keratectomy”. Am. J. Ophthalmol. 140 (1): 83–7. doi:10.1016/j.ajo.2005.02.037.PMID 15953577.
- Park-Wyllie, Laura Y.; David N. Juurlink; Alexander Kopp; Baiju R. Shah; Therese A. Stukel; Carmine Stumpo; Linda Dresser; Donald E. Low; Muhammad M. Mamdani (March 2006).“Outpatient Gatifloxacin Therapy and Dysglycemia in Older Adults”. The New England Journal of Medicine 354 (13): 1352–1361. doi:10.1056/NEJMoa055191. PMID 16510739. Retrieved 2006-05-01. Note: publication date 30 March; available on-line 1 March
- Gurwitz, Jerry H. (March 2006). “Serious Adverse Drug Effects — Seeing the Trees through the Forest”. The New England Journal of Medicine 354 (13): 1413–1415.doi:10.1056/NEJMe068051. PMID 16510740. Retrieved2006-05-01.
- Lewis-Hall, Freda (February 15, 2006). “Dear Healthcare Provider:” (PDF). Bristol-Myers Squibb. Retrieved May 1, 2006.
- Schmid, Randolph E. (May 1, 2006). “Drug Company Taking Tequin Off Market”. Associated Press. Archived from the original on November 25, 2007. Retrieved 2006-05-01.[dead link]
- “Two drugs banned”. The Hindu (Chennai, India). 19 March 2011.
- Peggy Peck (2 May 2006). “Bristol-Myers Squibb Hangs No Sale Sign on Tequin”. Med Page Today. Retrieved 24 February2009.
| EP0610958A2 * | 20 Jul 1989 | 17 Aug 1994 | Ube Industries, Ltd. | Intermediates in the preparation of 4-oxoquinoline-3-carboxylic acid derivatives |
| ES2077490A1 * | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008126384A1 | 31 Mar 2008 | 23 Oct 2008 | Daiichi Sankyo Co Ltd | Method for producing quinolone carboxylic acid derivative |
| CN101659654B | 28 Aug 2008 | 6 Nov 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | 18 Aug 2011 | 15 Feb 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
| EP1832587A1 * | 2 Mar 2007 | 12 Sep 2007 | Quimica Sintetica, S.A. | Method for preparing moxifloxacin and moxifloxacin hydrochloride |
| US7365201 | 2 Mar 2006 | 29 Apr 2008 | Apotex Pharmachem Inc. | Process for the preparation of the boron difluoride chelate of quinolone-3-carboxylic acid |
| US7875722 | 30 Sep 2009 | 25 Jan 2011 | Daiichi Sankyo Company, Limited | Method for producing quinolone carboxylic acid derivative |
| EP0464823A1 * | Jul 4, 1991 | Jan 8, 1992 | Kyorin Pharmaceutical Co., Ltd. | (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture |
| US4997943 * | Mar 31, 1987 | Mar 5, 1991 | Sankyo Company Limited | Quinoline-3-carboxylic acid derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101659654B | Aug 28, 2008 | Nov 6, 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | Aug 18, 2011 | Feb 15, 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
TAKE A TOUR
TAKE A TOUR
Amritsar, punjab, India
-
Amritsar – Wikipedia, the free encyclopedia
https://en.wikipedia.org/?title=AmritsarAmritsar is one of the largest cities of the Punjab state in India. The city origin lies in the village of Tung, and was named after the lake founded by the fourth Sikh …

 GOLDEN TEMPLE
GOLDEN TEMPLE

 .
.





 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar

Bullet marks on the walls of the park premises







The Jallianwalla Bagh in 1919, months after the massacre
Mealtime at the Golden Temple Amritsar
 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone
Sri Guru Ram Dass Jee International Airport in Amritsar
Amritsar – Wagah Border – Street food stall | Explore bernic… |

-

Golden Temple
-

Maharaja Ranjit Singh’s Ram Bagh Gardens
-

Golden Temple
-

Durgiana Temple
-

The holy water
-

Jallianwala Bagh
-

Jallianwala Bagh
-

The holy water
-

Golden Temple
-

Golden Temple
-

Sikh Gurdwara
-

The holy water
 Night view of the Harmandir Sahib///////////
Night view of the Harmandir Sahib/////////// -



%27,_oil_on_canvas_painting_by_Charles_W._Bartlett,_1920,_Honolulu_Academy_of_Arts.jpg)
 tandoori chicken
tandoori chicken












