OSILODROSTAT
LCI 699, LCI 699NX
Novartis Ag INNOVATOR
UNII-5YL4IQ1078, CAS 928134-65-0
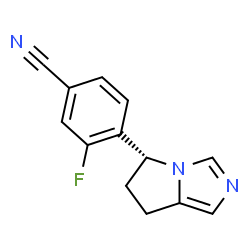
Benzonitrile, 4-[(5R)-6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl]-3-fluoro-
4-[(5R)-6,7-Dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl]-3-fluorobenzonitrile
(R)-4-(6,7-Dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-3-fluoro- benzonitrile
- Molecular FormulaC13H10FN3
- Average mass227.237 Da
- Originator Novartis
- Class Antihypertensives; Fluorobenzenes; Imidazoles; Nitriles; Pyridines; Small molecules
- Mechanism of Action Aldosterone synthase inhibitors
- Phase III Cushing syndrome
- Phase I Liver disorders
- Discontinued Heart failure; Hypertension; Solid tumours
Most Recent Events
- 27 Feb 2016 Novartis plans the phase III LINC-4 trial for Cushing’s syndrome in Greece, Thailand, Poland, Turkey, Russia, Brazil, Belgium, Spain, Denmark, Switzerland and USA (PO) (NCT02697734)
- 12 Jun 2015 Novartis plans a phase II trial for Cushing syndrome in Japan (NCT02468193)
- 01 Apr 2015 Phase-I clinical trials in Liver disorders in USA (PO)

Osilodrostat phosphate
CAS: 1315449-72-9
MF, C13-H10-F-N3.H3-O4-P
MW, 325.2347
Aromatase inhibitor; Cytochrome P450 11B1 inhibitor
MORE SYNTHESIS COMING, WATCH THIS SPACE…………………..

SYNTHESIS
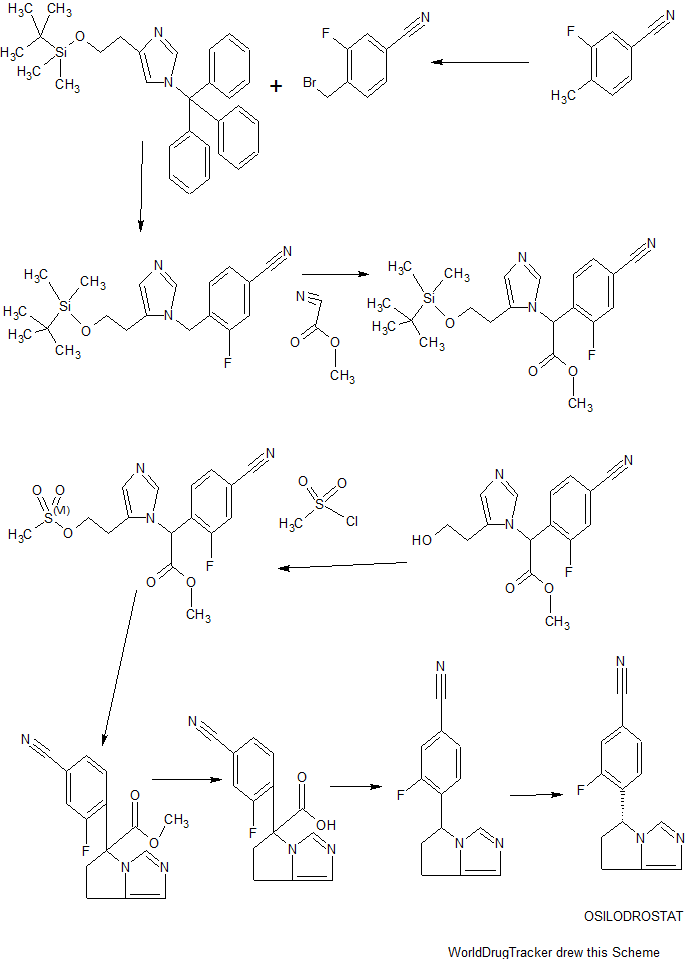
ACS Medicinal Chemistry Letters, 4(12), 1203-1207; 2013
REMIND ME, amcrasto@gmail.com, +919323115463
Osilodrostat, as modulators of 11-β-hydroxylase, useful for treating a disorder ameliorated 11-β-hydroxylase inhibition eg Cushing’s disease, hypertension, congestive heart failure, metabolic syndrome, liver diseases, cerebrovascular diseases, migraine headaches, osteoporosis or prostate cancer.
Novartis is developing osilodrostat, an inhibitor of aldosterone synthase and aromatase, for treating Cushing’s disease. In July 2016, osilodrostat was reported to be in phase 3 clinical development.
The somatostatin analog pasireotide and the 11β-hydroxylase inhibitor osilodrostat (LCI699) reduce cortisol levels by distinct mechanisms of action. There exists a scientific rationale to investigate the clinical efficacy of these two agents in combination. This manuscript reports the results of a toxicology study in rats, evaluating different doses of osilodrostat and pasireotide alone and in combination. Sixty male and 60 female rats were randomized into single-sex groups to receive daily doses of pasireotide (0.3mg/kg/day, subcutaneously), osilodrostat (20mg/kg/day, orally), osilodrostat/pasireotide in combination (low dose, 1.5/0.03mg/kg/day; mid-dose, 5/0.1mg/kg/day; or high dose, 20/0.3mg/kg/day), or vehicle for 13weeks. Mean body-weight gains from baseline to Week 13 were significantly lower in the pasireotide-alone and combined-treatment groups compared to controls, and were significantly higher in female rats receiving osilodrostat monotherapy. Osilodrostat and pasireotide monotherapies were associated with significant changes in the histology and mean weights of the pituitary and adrenal glands, liver, and ovary/oviduct. Osilodrostat alone was associated with adrenocortical hypertrophy and hepatocellular hypertrophy. In combination, osilodrostat/pasireotide did not exacerbate any target organ changes and ameliorated the liver and adrenal gland changes observed with monotherapy. Cmax and AUC0-24h of osilodrostat and pasireotide increased in an approximately dose-proportional manner. In conclusion, the pasireotide and osilodrostat combination did not exacerbate changes in target organ weight or toxicity compared with either monotherapy, and had an acceptable safety profile; addition of pasireotide to the osilodrostat regimen may attenuate potential adrenal gland hyperactivation and hepatocellular hypertrophy, which are potential side effects of osilodrostat monotherapy.

The somatostatin analog pasireotide and the 11β-hydroxylase inhibitor osilodrostat (LCI699) reduce cortisol levels by distinct mechanisms of action. There exists a scientific rationale to investigate the clinical efficacy of these two agents in combination. This manuscript reports the results of a toxicology study in rats, evaluating different doses of osilodrostat and pasireotide alone and in combination. Sixty male and 60 female rats were randomized into single-sex groups to receive daily doses of pasireotide (0.3 mg/kg/day, subcutaneously), osilodrostat (20 mg/kg/day, orally), osilodrostat/pasireotide in combination (low dose, 1.5/0.03 mg/kg/day; mid-dose, 5/0.1 mg/kg/day; or high dose, 20/0.3 mg/kg/day), or vehicle for 13 weeks. Mean body-weight gains from baseline to Week 13 were significantly lower in the pasireotide-alone and combined-treatment groups compared to controls, and were significantly higher in female rats receiving osilodrostat monotherapy. Osilodrostat and pasireotide monotherapies were associated with significant changes in the histology and mean weights of the pituitary and adrenal glands, liver, and ovary/oviduct. Osilodrostat alone was associated with adrenocortical hypertrophy and hepatocellular hypertrophy. In combination, osilodrostat/pasireotide did not exacerbate any target organ changes and ameliorated the liver and adrenal gland changes observed with monotherapy. Cmax and AUC0–24h of osilodrostat and pasireotide increased in an approximately dose-proportional manner.

In conclusion, the pasireotide and osilodrostat combination did not exacerbate changes in target organ weight or toxicity compared with either monotherapy, and had an acceptable safety profile; addition of pasireotide to the osilodrostat regimen may attenuate potential adrenal gland hyperactivation and hepatocellular hypertrophy, which are potential side effects of osilodrostat monotherapy.
The somatostatin class is a known class of small peptides comprising the naturally occurring somatostatin- 14 and analogues having somatostatin related activity, e.g. as disclosed by A.S. Dutta in Small Peptides, Vol.19, Elsevier (1993). By “somatostatin analogue” as used herein is meant any straight-chain or cyclic polypeptide having a structure based on that of the naturally occurring somatostatin- 14 wherein one or more amino acid units have been omitted and/or replaced by one or more other amino radical(s) and/or wherein one or more functional groups have been replaced by one or more other functional groups and/or one or more groups have been replaced by one or several other isosteric groups. In general, the term covers all modified derivatives of the native somatostatin- 14 which exhibit a somatostatin related activity, e.g. they bind to at least one of the five somatostatin receptor (SSTR), preferably in the nMolar range. Commonly known somatostatin analogs are octreotide, vapreotide, lanreotide, pasireotide.
Pasireotide, having the chemical structure as follow:
Pasireotide is called cyclo[{4-(NH2-C2H4-NH-CO-0-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)- Phe], wherein Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl, in free form, in salt or complex form or in protected form.
Cushing’s syndrome is a hormone disorder caused by high levels of Cortisol in the blood. This can be caused by taking glucocorticoid drugs, or by tumors that produce Cortisol or adrenocorticotropic hormone (ACTH) or CRH. Cushing’s disease refers to one specific cause of the syndrome: a tumor (adenoma) in the pituitary gland that produces large amounts of ACTH, which elevates Cortisol. It is the most common cause of Cushing’s syndrome, responsible for 70% of cases excluding glucocorticoid related cases. The significant decrease of Cortisol levels in Cushing’s disease patients on pasireotide support its potential use as a targeted treatment for Cushing’s disease (Colao et al. N Engl J Med 2012;366:32^12).
Compound A is potent inhibitor of the rate-limiting enzyme 1 1-beta-hydroxylase, the last step in the synthesis of Cortisol. WO 201 1/088188 suggests the potential use of compound A in treating a disease or disorder characterised by increased stress hormone levels and/or decreased androgen hormone levels, including the potential use of compound A in treating heart failure, cachexia, acute coronary syndrome, chronic stress syndrome, Cushing’s syndrome or metabolic syndrome.
Compound A, also called (R)-4-(6,7-Dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl)-3-fluoro- benzonitrile, has formula (II).
Compound A can be synthesized or produced and characterized by methods as described in WO2007/024945.
PRODUCT PATENT
WO2007024945, hold protection in the EU states until August 2026, and expire in the US in March 2029 with US154 extension
PAPER
ACS Medicinal Chemistry Letters (2013), 4(12), 1203-1207.
http://pubs.acs.org/doi/abs/10.1021/ml400324c?source=chemport&journalCode=amclct
Discovery and in Vivo Evaluation of Potent Dual CYP11B2 (Aldosterone Synthase) and CYP11B1 Inhibitors
Erik L. Meredith*†, Gary Ksander†, Lauren G. Monovich†, Julien P. N. Papillon†, Qian Liu†, Karl Miranda†, Patrick Morris†, Chang Rao†, Robin Burgis†, Michael Capparelli†, Qi-Ying Hu†, Alok Singh†, Dean F. Rigel‡, Arco Y. Jeng‡, Michael Beil‡, Fumin Fu‡, Chii-Whei Hu‡, and Daniel LaSala‡
† Novartis Institutes for BioMedical Research, 100 Technology Square, Cambridge, Massachusetts 02139, United States
‡ Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936, United States
ACS Med. Chem. Lett., 2013, 4 (12), pp 1203–1207
DOI: 10.1021/ml400324c
Aldosterone is a key signaling component of the renin-angiotensin-aldosterone system and as such has been shown to contribute to cardiovascular pathology such as hypertension and heart failure. Aldosterone synthase (CYP11B2) is responsible for the final three steps of aldosterone synthesis and thus is a viable therapeutic target. A series of imidazole derived inhibitors, including clinical candidate 7n, have been identified through design and structure–activity relationship studies both in vitro and in vivo. Compound 7n was also found to be a potent inhibitor of 11β-hydroxylase (CYP11B1), which is responsible for cortisol production. Inhibition of CYP11B1 is being evaluated in the clinic for potential treatment of hypercortisol diseases such as Cushing’s syndrome.
PATENT
WO-2016109361
silodrostat (LCI699; 4-[(5R)-6,7-dihydro-5H-pyrrolo[l,2-c]imidazol-5-yl]-3-fluoro-benzonitrile; CAS# 928134-65-0). Osilodrostat is a Ι Ι-β-hydroxylase inhibitor.
Osilodrostat is currently under investigation for the treatment of Cushing’s disease, primary aldosteronism, and hypertension. Osilodrostat has also shown promise in treating drug-resistant hypertension, essential hypertension, hypokalemia, hypertension, congestive heart failure, acute heart failure, heart failure, cachexia, acute coronary syndrome, chronic stress syndrome, Cushing’s syndrome, metabolic syndrome, hypercortisolemia, atrial fibrillation, renal failure, chronic renal failure, restenosis, sleep apnea, atherosclerosis, syndrome X, obesity, nephropathy, post-myocardial infarction, coronary heary disease, increased formation of collagen, cardiac or myocardiac fibrosis and/or remodeling following hypertension and endothelial dysfunction, Conn’s disease, cardiovascular diseases, renal dysfunction, liver diseases, cerebrovascular diseases, vascular diseases, retinopathy, neuropathy, insulinopathy, edema, endothelial dysfunction, baroreceptor dysfunction, migraine headaches, arrythmia, diastolic dysfunction, diastolic heart failure, impaired diastolic filling, systolic dysfunction, ischemia, hypertrophic cardiomyopathy, sudden cardia death, impaired arterial compliance, myocardial necrotic lesions, vascular damage, myocardial infarction, left ventricular hypertrophy, decreased ej ection fraction, cardiac lesions, vascular wall hypertrophy, endothelial thickening, fibrinoid, necrosis of coronary arteries, ectopic ACTH syndrome, change in adrenocortical mass, primary pigmented nodular adrenocortical disease (PPNAD), Carney complex (CNC), anorexia nervosa, chronic alcoholic poisoning, nicotine withdrawal syndrome, cocaine withdrawal syndrome, posttraumatic stress syndrome, cognitive impairment after a stroke or cortisol-induced mineral corticoid excess, ventricular arrythmia, estrogen-dependent disorders, gynecomastia, osteoporosis, prostate cancer, endometriosis, uterine fibroids, dysfunctional uterine bleeding, endometrial hyperplasia, polycyctic ovarian disease, infertility, fibrocystic breast disease, breast cancer, and fibrocystic mastopathy. WO 2013109514; WO 2007024945; and WO 2011064376.

Osilodrostat
Osilodrostat is likely subject to extensive CYP45o-mediated oxidative metabolism. These, as well as other metabolic transformations, occur in part through polymorphically-expressed enzymes, exacerbating interpatient variability. Additionally, some metabolites of osilodrostat derivatives may have undesirable side effects. In order to overcome its short half-life, the drug likely must be taken several times per day, which increases the probability of patient incompliance and discontinuance. Adverse effects associated with osilodrostat include fatigue, nausea, diarrhea, headache, hypokalemia, muscle spasms, vomiting, abdominal discomfort, abdominal pain, arthralgia, arthropod bite, dizziness, increased lipase, and pruritis.
Scheme I

EXAMPLE 1
(R)-4-(6,7-dihvdro-5H-pyrrolo[l,2-elimidazol-5-yl)-3-fluorobenzonitrile
(osilodrostat)

[00144] 4-(bromomethyl)-3-fluorobenzonitrile: 3-Fluoro-4-methylbenzonitrile (40 g, 296 mmol), NBS (63.2 g, 356 mmol) and benzoyl peroxide (3.6 g, 14.8 mmol) were taken up in carbon tetrachloride (490 mL) and refiuxed for 16 h. The mixture was allowed to cool to room temperature and filtered. The filtrate was concentrated and purified via flash column chromatography (0-5% EtOAc/hexanes) to give 4-(bromomethyl)-3-fluorobenzonitrile (35.4 g, 56%).

[00145] 2-(l-trityl-lH-imidazol-4-yl)acetic acid: Trityl chloride (40 g, 143.88 mmol, 1.2 equiv) was added to a suspension of (lH-imidazol-4-yl) acetic acid hydrochloride (20 g, 123.02 mmol, 1.0 equiv) in pyridine (200 mL). This was stirred at 50 °C for 16 h. Then the mixture was cooled and concentrated under vacuum and the crude product was purified by recrystallization from ethyl acetate (1000 ml) to afford 42 g (90%) of 2-[l-(triphenylmethyl)-lH-imidazol-4-yl] acetic acid as an off-white solid. LCMS (ESI): m/z = 369.2 [M+H]+
Step 2

2 step 2
2-( 1 -trityl- lH-imidazol-4-yl)ethanol : 2-(l-Trityl-lH-imidazol-4-yl) acetic acid (42 g, 114.00 mmol, 1.0 equiv) was suspended in THF (420 mL) and cooled to 0 °C. To this was added BH3 (1M in THF, 228.28 mL, 2.0 equiv). The clear solution obtained was stirred at 0 °C for 60 min, then warmed to room temperature until LCMS indicated completion of the reaction. The solution was cooled again to 0 °C and quenched carefully with water (300 mL). The resulting solution was extracted with ethyl acetate (3 x 100 mL) and the organic layers combined and dried over anhydrous Na2S04 and evaporated to give a sticky residue which was taken up in ethanolamine (800 mL) and heated to 90 °C for 2 h. The reaction was transferred to a separatory funnel, diluted with EtOAc (1 L) and washed with water (3 x 600 mL). The organic phase was dried over anhydrous Na2S04 and evaporated afford 35 g (87%) of 2-[l-(triphenylmethyl)-lH-imidazol-4-yl]ethanol as a white solid, which was used in the next step without further purification. LCMS (ESI) : m/z = 355.1 [M+H]+.
Step 3

3 step 3 4
4-(2-(tert-butyldimethylsilyloxy)ethyl)-l-trityl-lH-imidazole: 2-(l-Trityl-lH-imidazol-4-yl) ethanol (35 g, 98.75 mmol, 1.00 equiv) was dissolved in DCM (210 mL). To this was added imidazole (19.95 g, 293.05 mmol, 3.00 equiv) and tert-butyldimethylsilylchloride (22.40 g, 149.27 mmol, 1.50 equiv). The mixture was stirred at room temperature until LCMS indicated completion of the reaction. Then the resulting solution was diluted with 500 mL of DCM. The resulting mixture was washed with water (3 x 300 mL). The residue was purified by a silica gel column, eluted with ethyl
acetate/petroleum ether (1 :4) to afford 40 g (77%) of 4-[2-[(tert-butyldimethylsilyl)oxy]ethyl]-l-(triphenylmethyl)-lH-imidazole as a white solid. LCMS (ESI) : m/z = 469.1 [M+H]+.
Step 4

4-((5-(2-(tert-butyldimethylsilyloxy )ethylVlH-iniidazol-l -vnmethylV3-fluorobenzonitrile: 4-(2-((tert-Butyldimethylsilanyl)oxy)ethyl)-l rityl-lH-irnidazole (40 g, 85.34 mmol, 1.00 equiv) and 4-(Bromomethyl)-3-fluorobenzonitrile (27.38 g, 127.92 mmol, 1.50 equiv) obtained as a product of step 0, were dissolved in MeCN (480 mL) and DCM (80 mL), and stirred at room temperature for 48 h. Et2NH (80 mL) and MeOH (480 mL) were then added and the solution was warmed 80 °C for 3 h. The solution was evaporated to dryness and the residue was purified via flash column chromatography (EtOAc/hexanes 1 :5 to EtOAc) to afford 4-((5-(2-((tert-Butyldimethylsilanyl)oxy)ethyl)-lH-imidazol-l -yl)methyl)-3-fluorobenzonitrile (15 g, 50%). ¾ NMR (400 MHz, CDCh) δ: 7.67 (s, 1H), 7.43 (m, 2H), 6.98 (s, 1H), 6.88-6.79 (m, 1H), 5.34 (s, 2H), 3.79 (t, J= 8.0 Hz, 2H), 2.67 (t, J = 8.0 Hz, 2H), 0.88 (s, 9H), 0.02 (s, 6H). LCMS (ESI) : m/z = 360.1 [M+H]+.
Step 5

5 6
Methyl 2-(5-(2-(tert-butyldimethylsilyloxy)ethyl)-lH-imidazol-l -yl)-2-(4-cvano-2-fluorophenvDacetate: 4-((5-(2-((tert-Butyldimethylsilanyl)oxy)ethyl)-lH-imidazol-l -yl)methyl)-3-fluorobenzonitrile (15 g, 41.72 mmol, 1.00 equiv) was dissolved in anhydrous THF (150 mL) and stirred at -78 °C, then a THF solution of LiHMDS (75 mL, 1.80 equiv, 1.0 M) was added dropwise over 15 min. After 30 min, methyl cyanoformate (4.3 g, 45.50 mmol, 1.10 equiv) was added dropwise over 10 min and the solution was stirred at -78 °C for 2 h. The excess LiHMDS was quenched with aqueous saturated NH4CI and the mixture was allowed to warm to room temperature. The mixture was then diluted with EtOAc and washed
with aqueous saturated NH4CI (200 mL). The organic layers was dried over anhydrous Na2S04 and evaporated. The crude residue was purified via flash column chromatography (EtOAc/PE 3: 10 to EtOAc) to give methyl 2-(5-(2-((tert-butyldimethylsilanyl)oxy)ethyl)-lH-imidazol-l-yl)-2-(4-cyano-2-fluorophenyl) acetate (15 g, 86%) as a light yellow solid.
¾ NMR (400 MHz, CDCL3) δ: 7.66 (s, 1H), 7.54-7.43 (m, 2H), 7.15 (t, J= 8.0 Hz 1H), 6.93 (s, 1H), 6.47 (s, 1H), 3.88-3.74 (m, 5H), 2.81-2.62 (m, 2H), 0.89 (s, 9H), 0.05 (s, 6H) . LCMS (ESI) : m/z = 418.2 [M+H]+.
Step 6

Methyl 2-(4-cvano-2-fluorophenyl)-2-(5-(2-hvdroxyethyl)-lH-imidazol-l-yl) acetate: Methyl 2-(5-(2-((tert-butyldimethylsilanyl)oxy)ethyl)-lH-imidazol-l-yl)-2-(4-cyano-2-fiuorophenyl)acetate (15 g, 35.92 mmol, 1.00 equiv) was added to a solution of HCl in 1,4-dioxane (89 mL, 4.0 M, 359.2 mmol) at 0 °C and the mixture was allowed to warm to room temperature and stirred for 2 h. The solution was concentrated to dryness to give the crude alcohol, methyl 2-(4-cyano-2-fluorophenyl )-2-(5-(2 -hydroxy ethyl)-lH-imidazol-l-yl)acetate (10 g, 92%), which was used without further purification. LCMS: m/z = 304.0 [M+H]+.
Step 7

7 8
Methyl 2-(4-cvano-2-fluorophenyl)-2-(5-(2-(methylsulfonyloxy)ethyl)-lH-imidazol-l-yl) acetate: The crude methyl 2-(4-cyano-2-fluorophenyl )-2-(5-(2-hydroxyethyl)-lH-imidazol-l-yl)acetate (10 g, 32.97 mmol, 1.00 equiv) was dissolved in DCM (200 mL) and stirred at 0 °C, then Et3N (20 g, 197.65 mmol, 6.00 equiv) and
methanesulfonyl chloride (4.52 g, 39.67 mmol, 1.20 equiv) were added. After completion of the reaction, the solution was diluted with DCM and washed with aqueous saturated
NaHCC . The organic layer was dried over anhydrous Na2S04, filtered and evaporated to give the crude methyl 2-(4-cyano-2-fluorophenyl)-2-(5-(2-((methylsulfonyl)oxy)ethyl)-lH-imidazol-l-yl)acetate (11.43 g, 91%), which was used in the next step without further purification. LCMS (ESI) : m/z = 382.0 [M+H]+.
Step 8

Methyl 5-(4-cvano-2-fluorophenyl)-6.7-dihvdro-5H-pyrrolo[1.2-elimidazole-5-carboxylate: The crude methyl 2-(4-cyano-2 -fluorophenyl )-2-(5-(2- ((methylsulfonyl)oxy)ethyl)-lH-imidazol-l-yl)acetate (11.43 g, 29.97 mmol, 1.00 equiv) was dissolved in MeCN (550 mL) and then K2CO3 (12.44 g, 90.01 mmol, 3.00 equiv), Nal (13.50 g, 90.00 mmol, 3.00 equiv) and Et3N (9.09 g, 89.83 mmol, 3.00 equiv) were added. The reaction was stirred at 80 °C for 42 h. The mixture was filtered. The solids were washed with DCM. The filtrate was concentrated and purified by flash column chromatography (EtOAc) to give methyl 5-(4-cyano-2-fluorophenyl)-6,7-dihydro-5H-pyrrolo[l,2-c]imidazole-5-carboxylate (4.2 g, 49% in 3 steps).
[00153] ¾ NMR (400 MHz, CDCb) δ: 7.61 (s, 1H), 7.47-7.47 (m, 2H), 6.88 (s, 1H), 6.79-6.75 (m, 1H), 4.17-4.12 (m, 1H), 3.87 (s, 3H), 3.78-3.70 (m, 1H), 3.08-3.02 (m, 1H), 2.84-2.71 (m, 2H). LCMS (ESI) : m/z = 286.0 [M+H]+.
Step 9

10
4-(6.7-dihvdro-5H-pyrrolo[1.2-elimidazol-5-yl)-3-fluorobenzonitrile: To a 40-mL sealed tube, was placed methyl 5-(4-cyano-2-fluorophenyl)-5H,6H,7H-pyrrolo[l,2-c]imidazole-5-carboxylate (1 g, 3.51 mmol, 1.00 equiv), DMSO (10 mL), water (5 mL). The final reaction mixture was irradiated with microwave radiation for 40 min at 140 °C. The resulting solution was diluted with 100 mL of EtOAc. The resulting mixture was washed with (3 x 20 mL) brine, dried over anhydrous Na2S04, filtered and concentrated. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (4: 1) to afford 420 mg (44%) of 5-(4-cyano-2-fluorophenyl)-5H,6H,7H-pyrrolo[l,2-c]irnidazole-5-carboxylic acid as a light yellow solid.
¾ NMR (400 MHz, CDCL3) δ: 7.55-7.28 (m, 3H), 6.90-6.85 (m, 2H), 5.74-5.71 (m, 1H), 3.25-3.15 (m, 1H), 3.02-2.92 (m, 2H), 2.58-2.50 (m, 1H). LCMS (ESI) : m/z = 228.2 [M+H]+.
Step 10

10
(R)-4-(6 -dihvdro-5H-pyrrolo[1.2-elirnidazol-5-yl)-3-fluorobenzonitrile:
Resolution of the enantiomers of the title compound (300 mg) was performed by chiral HPLC: Column, Chiralpak IA2, 2*25cm, 20um; mobile phase, Phase A: Hex (50%, 0.1% DEA), Phase B: EtOH (50%) ; Detector, UV 254/220 nm to afford the (S)-enantiomer (RT = 17 min) and the (R)-enantiomer (97.6 mg, desired compound) (RT = 21 min).
¾ NMR (400 MHz, DMSO-<4) δ: 7.98-7.95 (m, 1H), 7.70-7.69 (m, 1H), 7.50 (s, 1H), 6.87 (t, J= 8.0 Hz, 1H), 6.70 (s, 1H), 5.79-5.76 (m, 1H), 3.15-3.06 (m, 1H), 2.92-2.74 (m, 2H), 2.48-2.43 (m, 1H). LCMS (ESI) : m/z = 228.1 [M+H]+.
PATENT
WO2013/153129
https://www.google.com/patents/WO2013153129A1?cl=en
PATENT
WO2007/024945
http://www.google.co.in/patents/WO2007024945A1?cl=en
PATENT
EP 2815749
Aspect (iii) of the present invention relates to phosphate salt or nitrate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile according to Formula (III)

abbreviated as ‘{drug3}’. In particular, the present invention relates to crystalline form of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3a}’; to crystalline Form A of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3b}’; to crystalline Form B of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3c}’; to crystalline Form C of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3d}’; to crystalline Form D of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3e}’; to crystalline Form E of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3f}’; to crystalline Form F of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3g}’; to crystalline Form G of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3h}’; to crystalline Form H of phosphate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3i}’; and to crystalline form of nitrate salt of 4-(R)-(6,7-dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl)-3-fluoro-benzonitrile, abbreviated as ‘{drug3j}’. {drug3a}, {drug3b}, {drug3c}, {drug3d}, {drug3e}, {drug3f}, {drug3g}, {drug3h}, {drug3i}, and {drug3j} are specific forms falling within the definition of {drug3}. Aspect (iii) of the invention is separate from aspects (i), (ii), (iv), (v), (vi), (vii), and (viii) of the invention. Thus, all embodiments of {drug3a}, {drug3b}, {drug3c}, {drug3d}, {drug3e}, {drug3f}, {drug3g}, {drug3h}, {drug3i}, and {drug3j}, respectively, are only related to {drug3}, but neither to {drug1}, nor to {drug2}, nor to {drug4}, nor to {drug5}, nor to {drug6}, nor to {drug7}, nor to {drug8}.
PAPER
Osilodrostat (LCI699), a potent 11β-hydroxylase inhibitor, administered in combination with the multireceptor-targeted somatostatin analog pasireotide: A 13-week study in rats
- Li Lia, , ,
- Kapil Vashishta,
- Julie Boisclaira,
- Wenkui Lib,
- Tsu-han Linb,
- Herbert A. Schmidc,
- William Kluwea,
- Heidi Schoenfelda,
- Peter Hoffmanna
- a Preclinical Safety, Novartis Institutes for BioMedical Research, East Hanover, NJ, USA
- b Drug Metabolism and Pharmacokinetics, Novartis Institutes for BioMedical Research, East Hanover, NJ, USA
- c Novartis Oncology Development, Basel, Switzerland
doi:10.1016/j.taap.2015.05.004, http://www.sciencedirect.com/science/article/pii/S0041008X15001684
CLIPS


| WO2011088188A1 * |
Jan 13, 2011 |
Jul 21, 2011 |
Novartis Ag |
Use of an adrenal hormone-modifying agent |
REFERENCES
1: Guelho D, Grossman AB. Emerging drugs for Cushing’s disease. Expert Opin Emerg Drugs. 2015 Sep;20(3):463-78. doi: 10.1517/14728214.2015.1047762. Epub 2015 Jun 2. PubMed PMID: 26021183.
2: Li L, Vashisht K, Boisclair J, Li W, Lin TH, Schmid HA, Kluwe W, Schoenfeld H, Hoffmann P. Osilodrostat (LCI699), a potent 11β-hydroxylase inhibitor, administered in combination with the multireceptor-targeted somatostatin analog pasireotide: A 13-week study in rats. Toxicol Appl Pharmacol. 2015 Aug 1;286(3):224-33. doi: 10.1016/j.taap.2015.05.004. Epub 2015 May 14. PubMed PMID: 25981165.
3: Papillon JP, Adams CM, Hu QY, Lou C, Singh AK, Zhang C, Carvalho J, Rajan S, Amaral A, Beil ME, Fu F, Gangl E, Hu CW, Jeng AY, LaSala D, Liang G, Logman M, Maniara WM, Rigel DF, Smith SA, Ksander GM. Structure-Activity Relationships, Pharmacokinetics, and in Vivo Activity of CYP11B2 and CYP11B1 Inhibitors. J Med Chem. 2015 Jun 11;58(11):4749-70. doi: 10.1021/acs.jmedchem.5b00407. Epub 2015 May 21. PubMed PMID: 25953419.
4: Fleseriu M. Medical treatment of Cushing disease: new targets, new hope. Endocrinol Metab Clin North Am. 2015 Mar;44(1):51-70. doi: 10.1016/j.ecl.2014.10.006. Epub 2014 Nov 4. Review. PubMed PMID: 25732642.
5: Wang HZ, Tian JB, Yang KH. Efficacy and safety of LCI699 for hypertension: a meta-analysis of randomized controlled trials and systematic review. Eur Rev Med Pharmacol Sci. 2015;19(2):296-304. Review. PubMed PMID: 25683946.
6: Daniel E, Newell-Price JD. Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing’s syndrome. Eur J Endocrinol. 2015 Jun;172(6):R263-80. doi: 10.1530/EJE-14-1014. Epub 2015 Jan 30. Review. PubMed PMID: 25637072.
7: Fleseriu M, Petersenn S. Medical therapy for Cushing’s disease: adrenal steroidogenesis inhibitors and glucocorticoid receptor blockers. Pituitary. 2015 Apr;18(2):245-52. doi: 10.1007/s11102-014-0627-0. PubMed PMID: 25560275.
8: Ménard J, Rigel DF, Watson C, Jeng AY, Fu F, Beil M, Liu J, Chen W, Hu CW, Leung-Chu J, LaSala D, Liang G, Rebello S, Zhang Y, Dole WP. Aldosterone synthase inhibition: cardiorenal protection in animal disease models and translation of hormonal effects to human subjects. J Transl Med. 2014 Dec 10;12:340. doi: 10.1186/s12967-014-0340-9. PubMed PMID: 25491597; PubMed Central PMCID: PMC4301837.
9: Oki Y. Medical management of functioning pituitary adenoma: an update. Neurol Med Chir (Tokyo). 2014;54(12):958-65. Epub 2014 Nov 29. PubMed PMID: 25446388.
10: Cai TQ, Stribling S, Tong X, Xu L, Wisniewski T, Fontenot JA, Struthers M, Akinsanya KO. Rhesus monkey model for concurrent analyses of in vivo selectivity, pharmacokinetics and pharmacodynamics of aldosterone synthase inhibitors. J Pharmacol Toxicol Methods. 2015 Jan-Feb;71:137-46. doi: 10.1016/j.vascn.2014.09.011. Epub 2014 Oct 7. PubMed PMID: 25304940.
11: Lother A, Moser M, Bode C, Feldman RD, Hein L. Mineralocorticoids in the heart and vasculature: new insights for old hormones. Annu Rev Pharmacol Toxicol. 2015;55:289-312. doi: 10.1146/annurev-pharmtox-010814-124302. Epub 2014 Sep 10. Review. PubMed PMID: 25251996.
12: Cuevas-Ramos D, Fleseriu M. Treatment of Cushing’s disease: a mechanistic update. J Endocrinol. 2014 Nov;223(2):R19-39. doi: 10.1530/JOE-14-0300. Epub 2014 Aug 18. Review. PubMed PMID: 25134660.
13: Yin L, Hu Q, Emmerich J, Lo MM, Metzger E, Ali A, Hartmann RW. Novel pyridyl- or isoquinolinyl-substituted indolines and indoles as potent and selective aldosterone synthase inhibitors. J Med Chem. 2014 Jun 26;57(12):5179-89. doi: 10.1021/jm500140c. Epub 2014 Jun 5. PubMed PMID: 24899257.
14: Li W, Luo S, Rebello S, Flarakos J, Tse FL. A semi-automated LC-MS/MS method for the determination of LCI699, a steroid 11β-hydroxylase inhibitor, in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2014 Jun 1;960:182-93. doi: 10.1016/j.jchromb.2014.04.012. Epub 2014 Apr 30. PubMed PMID: 24814004.
15: Trainer PJ. Next generation medical therapy for Cushing’s syndrome–can we measure a benefit? J Clin Endocrinol Metab. 2014 Apr;99(4):1157-60. doi: 10.1210/jc.2014-1054. PubMed PMID: 24702012.
16: Bertagna X, Pivonello R, Fleseriu M, Zhang Y, Robinson P, Taylor A, Watson CE, Maldonado M, Hamrahian AH, Boscaro M, Biller BM. LCI699, a potent 11β-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab. 2014 Apr;99(4):1375-83. doi: 10.1210/jc.2013-2117. Epub 2013 Dec 11. PubMed PMID: 24423285.
17: Oki Y. Medical management of functioning pituitary adenoma: an update. Neurol Med Chir (Tokyo). 2014;54 Suppl 3:958-65. PubMed PMID: 26236804.
18: Schumacher CD, Steele RE, Brunner HR. Aldosterone synthase inhibition for the treatment of hypertension and the derived mechanistic requirements for a new therapeutic strategy. J Hypertens. 2013 Oct;31(10):2085-93. doi: 10.1097/HJH.0b013e328363570c. PubMed PMID: 24107737; PubMed Central PMCID: PMC3771574.
19: Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. 2013 Aug;9(8):459-69. doi: 10.1038/nrneph.2013.110. Epub 2013 Jun 18. Review. PubMed PMID: 23774812; PubMed Central PMCID: PMC3922409.
20: van der Pas R, de Herder WW, Hofland LJ, Feelders RA. Recent developments in drug therapy for Cushing’s disease. Drugs. 2013 Jun;73(9):907-18. doi: 10.1007/s40265-013-0067-6. Review. PubMed PMID: 23737437.
///////OSILODROSTAT, Novartis , osilodrostat, an inhibitor of aldosterone synthase and aromatase, treating Cushing’s disease, July 2016, phase 3 clinical development, LCI 699, 928134-65-0, 1315449-72-9, PHASE 3, LCI 699NX, LCI 699AZA, CYP11B1 CYP11B2
c1cc(c(cc1C#N)F)[C@H]2CCc3n2cnc3.OP(=O)(O)O
N#CC1=CC=C([C@H]2CCC3=CN=CN32)C(F)=C1












 .
. Corresponding author email hreissig@chemie.fu-berlin.de
Corresponding author email hreissig@chemie.fu-berlin.de![[1860-5397-12-118-i1]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-12-118-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
 .
.

