
WO-2016092561, Ivacaftor, NEW PATENT
https://www.google.com/patents/WO2016092561A2?cl=en
Novel polymorphs of ivacaftor, process for its preparation and pharmaceutical composition thereof
Laurus Labs Pvt Ltd
LAURUS LABS PRIVATE LIMITED [IN/IN]; Plot No. DS1, IKP Knowledge Park, Genome Valley Turkapally, Shameerpet Mandal, Ranga District Hyderabad 500078 (IN)

THAIMATTAM, Ram; (IN).
DAMA, Venkata Srinivasa Rao; (IN).
INDUKURI, Venkata Sunil Kumar; (IN).
GORANTLA, Seeta Rama Anjaneyulu; (IN).
CHAVA, Satyanarayana; (IN)

Novel crystalline forms of ivacaftor (designated as forms L1 to L14), processes for their preparation and composition comprising them are claimed.
Vertex, in research collaboration with Cystic Fibrosis Foundation Therapeutics, had developed and launched ivacaftor.
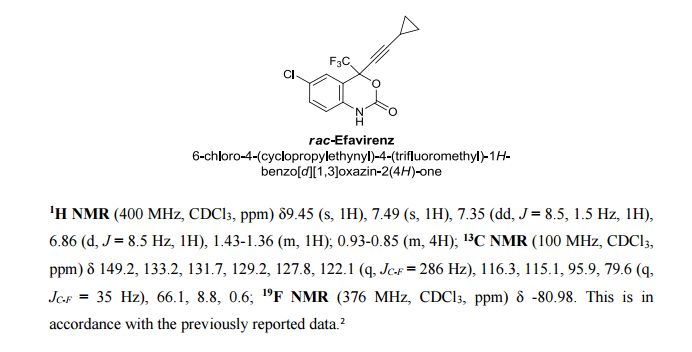
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula I:

Formula I
Ivacaftor was approved by FDA and marketed by Vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets.
WO2006/002421 publication discloses modulators of ATP-binding cassette transporters such as ivacaftor. This patent generally discloses a process for the preparation of modulators of ATP-binding cassette transporters such as quinoline compounds; however, specific process for the preparation of ivacaftor and its solid state details were not specifically disclosed.
WO2007/079139 publication discloses Form A, Form B and amorphous form of ivacaftor characterized by PXRD, DSC and TGA and process for their preparation. Further this publication discloses ethanol crystalate of ivacaftor in example part.
WO2009/038683 publication discloses the solid forms of ivacaftor, which are designated as Form-I (2-methylbutyric acid), Form-II (propylene glycol), Form-HI (PEG400.KOAc), Form-IV (lactic acid), Form-V (isobutyric acid), Form-VI (propionic
acid), Form- VII (ethanol), Form- VIII (2-propanol), Form-IX (monohydrate), Form-X (besylate Form A), Form-XI (besylate Form B), Form-XII (besylate Form D), Form-XIII (besylate Form E), Form-XIV (besylate Form F), Form-XV (besylate (2: 1)), Form-XVI (besylate mono hydrate). This publication also discloses the characterization details like PXRD, DSC and TGA for the above forms and process for their preparation.
WO201 1/1 16397 publication discloses crystalline Form C of ivacaftor, process for its preparation and pharmaceutical composition comprising the same. Also discloses characterization details of Form C, such as PXRD, IR, DSC and 13CSSNMR.
WO2013/158121 publication discloses solvated forms of ivacaftor, which are designated as Form D (acetonitrile or acetonitrile/water (75/25) solvate), Form E (Methyl ethyl ketone (MEK), MEK/water (90/1), MEK/water (90/10), MEK/water (80/20) solvate), Form F (acetonitrile/water (75/25) solvate), Form G (isopropyl acetate solvate), Form H (isopropyl acetate/water (95/5) solvate), Form I (MEK solvate), Form J (MEK/water (99/1) solvate), Form K (MEK or MEK/water (99/1) or MEK/water (90/10) or MEK/water (80/20) solvate), Form L (isopropyl acetate/water (95/5) solvate), Form M (MEK or MEK/water (99/1) solvate), Form N (MEK water (90/10) or MEK/water (80/20) solvate), Form O (MEK or MEK/water (99/1) solvate), Form P (MEK water (90/10) or MEK water (80/20) solvate), Form Q (MEK/water (80/20) solvate), Form R (acetonitrile solvate), Form S (MEK/water (80/20) solvate), Form T (isopropyl acetate/water (95/5) solvate), Form W (acetonitrile/water (90/10) solvate), Form XX (from 10% water/ acetonitrile) and hydrate B (hydrated form). This patent further discloses characterization details like PXRD and TGA for the above forms and process for their preparation.
WO2014/118805 publication discloses crystalline forms of ivacaftor designated as Form D, Form E, Form El, Form G and Form G’; amorphous ivacaftor designated as Form I and Form II; crystalline ivacaftor solvates such as n-butanol solvate, methanol solvate, propylene glycol solvate, DMF solvate, THF solvate, DMF:ethylacetate solvate. This publication further discloses the process for the preparation of said forms along with their characterization details.
WO2015/070336 publication discloses polymorphic form APO-I and MIBK solvate of ivacaftor along with its characteristic PXRD details, process for its preparation and pharmaceutical composition comprising them.
CN 104725314A publication discloses ivacaftor new polymorph D, which is obtained by crystallization of ivacaftor from acetonitrile/water. This publication further discloses characteristic details such PXRD, IR and DSC of ivacaftor new polymorph D.
Polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x-ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predictable solubility profiles. It is desirable to investigate all solid state forms of a drug, including all polymorphic forms and solvates, and to determine the stability, dissolution and flow properties of each polymorphic form.
Polymorphic forms and solvates of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry. Additionally, polymorphic forms and solvates of the same drug substance or active pharmaceutical ingredient, can be administered by itself or formulated as a drug product (also known as the final or finished dosage form), and are well known in the pharmaceutical art to affect, for example, the solubility, stability, flowability, tractability and compressibility of drug substances and the safety and efficacy of drug products.
The discovery of new polymorphic forms and solvates of a pharmaceutically useful compound, like ivacaftor, may provide a new opportunity to improve the performance characteristics of a pharmaceutical product. It also adds to the material that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic. New polymorphic forms of the ivacaftor have now been discovered and have been designated as ivacaftor Form-Ll, Form-L2, Form-L3, Form-L4, Form-L5, Form-L6, Form-L7, Form-L8, Form-L9, Form-LlO, Form-Ll 1, Form-Ll 2 A, Form-Ll 2B, Form-Ll 3 and Form-Ll 4.
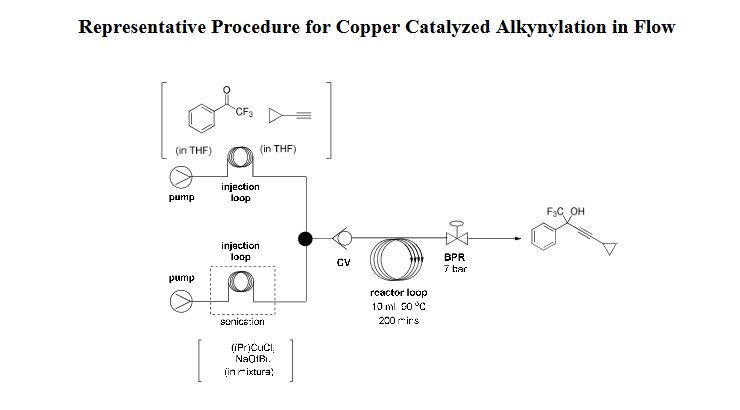
EXAMPLE 1 : Preparation of Ivacaftor Form-Ll
A suspension of ivacaftor ethanolate (5 g) in n-heptane (200 mL) was heated to 95-100°C and stirred for 5 hrs at the same temperature. Then the reaction mixture was cooled to 25-35°C and stirred for an hour. The solid obtained was filtered, washed with n-heptane and suck dried. The wet solid was further dried at 60-65°C for 16 hrs under vacuum yielded ivacaftor Form-Ll . The XRPD is set forth in Figure- 1.
In a similar manner, ivacaftor Form-Ll was prepared from different solvates of ivacaftor in place of ivacaftor ethanolate as input using the following conditions;

Ivacaftor cyclopentyl methyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr
Ivacaftor methyltertiarybutyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr

Dr Satyanarayana Chava
Chief executive officer (CEO)
When Dr Satyanarayana Chava started Laurus Labs in 2007, he invested nearly Rs 60 crore of his own money into it. His confidence in its success was neither bravado nor bluster, but defined by his knowledge of the pharmaceutical industry. Eight years on, the Hyderabad-based company is on track to reach revenues of Rs 2,000 crore by the end of FY2016.
Chava, now 52, has more than two decades of experience in the pharmaceutical industry; in his last job, he was chief operating officer (COO) of the successful startup, Matrix Laboratories. Of his 10 years there, he says with pride, “I never skipped a promotion and got to work in all departments.” His dedication, coupled with a sound understanding of what it takes to start a pharmaceutical company, is what makes Laurus Labs among the hottest startups in this sector.
Initially, Chava planned the business around research and development (R&D). He wanted Laurus Labs to focus on contract research and make money from royalties. “In India, companies start with manufacturing and then get into R&D,” he explains. “I did it the other way round.” He focussed his fledgling company’s resources on developing formulations for medicines, and licensed them to other pharmaceutical players. In the early months, Laurus Labs had 10 people in manufacturing and 300 in R&D.

In June 2007, Aptuit, a US-based contract research organisation (CRO), signed it on for a $20 million (then Rs 80 crore) contract. But despite this injection of funds, Chava was unable to sustain his original idea of developing technologies for other companies. At the time of the Aptuit deal, Laurus Labs’s annual revenues were not even $20,000 (Rs 8 lakh at the time). In 2008, Chava decided to start manufacturing active pharmaceutical ingredients (API), which, as the name suggests, are chemicals or key ingredients in drugs required to make the medication work. His early investment into R&D benefitted Laurus Labs; it maintains a large repository of research-based knowledge that forms the bedrock of any successful pharmaceutical business.
Today, it is a key manufacturer supplier of APIs and holds its own against better-known competitors like US generic drug giant Mylan, which, incidentally, acquired a controlling stake in Matrix around the time Chava founded Laurus Labs. It has also carved a niche for itself by supplying antiretroviral or ARVs (used to fight infections caused by retroviruses like HIV) and oncology drugs. And despite being a relatively new player, its clients include giants like Pfizer, Teva Pharmaceutical Industries and Merck.

The person behind it
A Master’s degree in chemistry was never on the cards for Chava. In the early 1980s, the best students usually studied physics, and he had planned to do the same. But when he went to his college in Amravati (Andhra Pradesh) to enroll, his elder sister’s friend suggested he study chemistry too. Chava took up the subject on a whim. He ended up liking chemistry so much so that in his final year he topped his batch despite not having written one out of the four required papers. He went on to complete his PhD in the subject in 1991.
Upon graduating, he was hired by Ranbaxy Laboratories in Delhi as a researcher. In those early years itself Chava knew he’d spend a lifetime in the industry. He enjoyed the work and gained valuable experience as a young researcher in what was then India’s finest pharmaceutical company.
But through his years in the industry, Chava was conscious of the fact that he needed to broaden his experience outside of research. His stint at Matrix Laboratories afforded him that opportunity. As it was a startup, he was able to rise through the ranks quickly and got the opportunity to work in key departments from sales and marketing to finance and accounts. Within eight years of joining Matrix, he became its COO.

This experience was to come in handy when, due to differences with the board—he refused to elaborate on this—he decided to leave Matrix and set up Laurus Labs. And though he is the company’s chief executive officer (CEO), Chava remains true to his calling as a chemist. He has strived to build an organisation that is not very hierarchical. It is not uncommon to see him interacting with the chemists in the company and discussing formulations with them—something unheard of in an industry where most CEOs are from a sales and marketing background.


Chandrakanth Chereddi
VP Synthesis Business Unit
Prior to his current assignment at Laurus Labs India, Chandra headed the Project Management division for all scientific projects at the Laurus R&D center. Chandra previously worked for McKinsey & Company in India as a member of the healthcare practice and at Google Inc. as a software engineer in Google’s Mountain View, CA office. Chandra holds a BE from the College of Engineering, Osmania University, Hyderabad, and MS from University of Illinois at Urbana-Champaign, and an MBA from Indian School of Business, Hyderabad.
///////WO 2016092561, Ivacaftor, New patent, Laurus Labs Pvt Ltd














.png)





1521-3773/asset/olalertbanner.jpg?v=1&s=0c0a7bae38bad177497650f0bc0083504684f4fa)
















{kind=link}