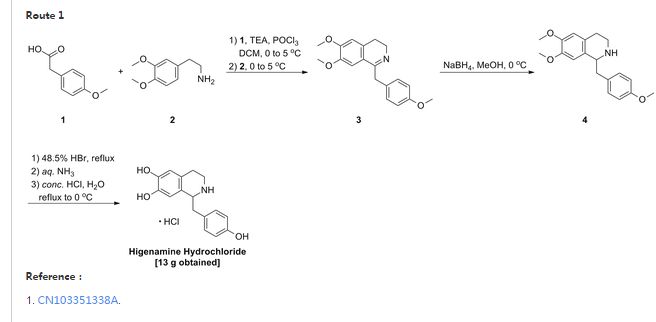
DSM265
DSM-265; PfSPZ
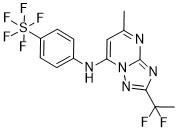
2-(1,1-difluoroethyl)-5-methyl-N-(4-(pentafluoro-l6-sulfanyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine
2-(l,l-difluoroethyl)-5-methyl-N-[4-(pentafluoro- 6– sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
(OC-6-21)-[4-[[2-(1,1-Difluoroethyl)-5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7-yl]amino]phenyl]pentafluorosulfur
1282041-94-4
Chemical Formula: C14H12F7N5S
Exact Mass: 415.0702
Board Of Regents, University Of Texas System, Monash University, Medicines For Malaria Venture
DSM265 is a long-duration, potent and selective dihydroorotate dehydrogenase (DHODH)) inhibitor. DSM265 is potential useful for the prevention and treatment of malaria. DSM265 is the first DHODH inhibitor to reach clinical development for treatment of malaria. DSM265 is highly selective toward DHODH of the malaria parasite Plasmodium, efficacious against both blood and liver stages of P. falciparum, and active against drug-resistant parasite isolates. DSM265 has advantages over current treatment options that are dosed daily or are inactive against the parasite liver stage.
- OriginatorMonash University; University of Texas Southwestern Medical Center; University of Washington
- Developer Center for Infectious Disease Research; Fred Hutchinson Cancer Research Center; Medicines for Malaria Venture; Takeda; United States Department of Defense
- Class Antimalarials; Pyrimidines; Small molecules; Triazoles
- Mechanism of Action Dihydroorotate dehydrogenase inhibitors
- Phase II Malaria
- Phase I Malaria
Most Recent Events
- 25 Apr 2016 Medicines for Malaria Venture and AbbVie plan a phase I bioavailability trial in Healthy volunteers in USA (PO, Granule) (NCT02750384)
- 01 Mar 2016 Phase-I clinical trials in Malaria prevention (In volunteers) in USA (PO) (NCT02562872)
- 01 Jan 2016 Phase-II clinical trials in Malaria in Peru (PO) (NCT02123290)
Malaria is one of the most significant causes of childhood mortality, but disease control efforts are threatened by resistance of the Plasmodium parasite to current therapies. Continued progress in combating malaria requires development of new, easy to administer drug combinations with broad-ranging activity against all manifestations of the disease. DSM265, a triazolopyrimidine-based inhibitor of the pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH), is the first DHODH inhibitor to reach clinical development for treatment of malaria. We describe studies profiling the biological activity, pharmacological and pharmacokinetic properties, and safety of DSM265, which supported its advancement to human trials. DSM265 is highly selective toward DHODH of the malaria parasite Plasmodium, efficacious against both blood and liver stages of P. falciparum, and active against drug-resistant parasite isolates. Favorable pharmacokinetic properties of DSM265 are predicted to provide therapeutic concentrations for more than 8 days after a single oral dose in the range of 200 to 400 mg. DSM265 was well tolerated in repeat-dose and cardiovascular safety studies in mice and dogs, was not mutagenic, and was inactive against panels of human enzymes/receptors. The excellent safety profile, blood- and liver-stage activity, and predicted long half-life in humans position DSM265 as a new potential drug combination partner for either single-dose treatment or once-weekly chemoprevention. DSM265 has advantages over current treatment options that are dosed daily or are inactive against the parasite liver stage.

A new single-dose malaria drug is offering promise as both a cure to malaria and also a way to prevent the disease according to researchers at UT Southwestern Medical Center. The new drug, which is known as DSM265, kills the drug-resistant malaria parasites in the blood and liver by targeting the ability of the parasites to replicate.

Malaria is a very infectious disease that is transmitted by mosquitoes, and it kills about 600,000 people worldwide every year. Most of the people who are killed by malaria are under 5-years-old, and it’s more common in sub-Saharan Africa. Almost 200 million cases of malaria are reported every year, with about 3 billion people in 97 countries at risk for the disease. Lead author Dr. Margaret Phillips, who is a professor of Pharmacology at UT Southwestern said that this could be the first single-dose cure for malaria, and would be used in partnership with another drug. This drug could also be developed into a once-a-week preventive vaccination as well, and the results of the study were just published in Science Translational Medicine. Not only was UT Southwestern involved in the research study, but Monash Institute of Pharmaceutical Sciences in Australia, the University of Washington, and the not-for-profit Medicines for Malaria Venture was also involved.
Malaria is one of the most deadly infectious diseases in human history with 3.2 billion people in 97 countries at risk. An estimated 444,000 deaths from malaria were reported by the WHO in 2015 and ∼90% of these occurred in sub-Saharan Africa, mostly among children under the age of five. Human malaria, which is transmitted by the female Anopheles mosquito, can be caused by five species of Plasmodia; however, Plasmodium falciparum and Plasmodium vivax are the most signficant.P. falciparum is dominant in Africa and accounts for most of the deaths, while P. vivax has a larger global distribution.
To simplify treatment options it is desirable that new drugs be efficacious against all human infective species. Malaria is a treatable disease and malarial control programs depend on drug therapy for treatment and chemoprevention, and on insecticides (including insecticide impregnated bed nets) to prevent transmission.
A large collection of drugs has been used for the treatment of malaria, but many of the most important compounds have been lost to drug resistance (e.g., chloroquine and pyrimethamine).Artemisinin combination therapies (ACT) replaced older treatments, becoming highly effective, crucial tools in global efforts that have led to the decline in malaria deaths over the past decade. However, resistance to the artemisinin components (associated with Kelch13 propeller protein mutations has been found in Southeast Asia putting at risk malaria treatment programs. To combat drug resistance a significant effort is underway to identify new compounds that can be used for the treatment of malaria, with several new entities currently in clinical development.

The triazolopyrimidine DSM265 developed by the group is the first antimalarial agent that targets dihydroorotate dehydrogenase (DHODH) to reach clinical development, validating this target for the treatment of malaria. DHODH is a mitochondrial enzyme that is required for the fourth step of de novo pyrimidine biosynthesis, catalyzing the flavin-dependent oxidation of dihydroorotate to orotic acid with mitochondrially derived coenzyme Q (CoQ) serving as a second substrate. Pyrimidines are essential for both RNA and DNA biosynthesis, and because Plasmodia do not encode pyrimidine salvage enzymes, which are found in humans and other organisms, the de novo pyrimidine pathway and DHODH are essential to the parasite.
They identified the triazolopyrimidine DHODH inhibitor series by a target-based high throughput screen, and the initial lead DSM1 (2) was shown to selectively inhibit P. falciparumDHODH and to kill parasites in vitro, but it was ineffective in vivo due to poor metabolic properties. The series was subsequently optimized to improve its in vivo properties resulting in the identification of DSM74 (3), which while metabolically stable lacked potencyX-ray structures of 2 and 3 bound to PfDHODH were then used to guide the medicinal chemistry program in the search for more potent analogues, resulting in the identification of 1.
SYNTHESIS
PAPER
Journal of Medicinal Chemistry (2012), 55(17)

Plasmodium falciparum causes approximately 1 million deaths annually. However, increasing resistance imposes a continuous threat to existing drug therapies. We previously reported a number of potent and selective triazolopyrimidine-based inhibitors of P. falciparum dihydroorotate dehydrogenase that inhibit parasite in vitro growth with similar activity. Lead optimization of this series led to the recent identification of a preclinical candidate, showing good activity against P. falciparum in mice. As part of a backup program around this scaffold, we explored heteroatom rearrangement and substitution in the triazolopyrimidine ring and have identified several other ring configurations that are active as PfDHODH inhibitors. The imidazo[1,2-a]pyrimidines were shown to bind somewhat more potently than the triazolopyrimidines depending on the nature of the amino aniline substitution. DSM151, the best candidate in this series, binds with 4-fold better affinity (PfDHODH IC50 = 0.077 μM) than the equivalent triazolopyrimidine and suppresses parasites in vivo in the Plasmodium berghei model.
Scheme 3
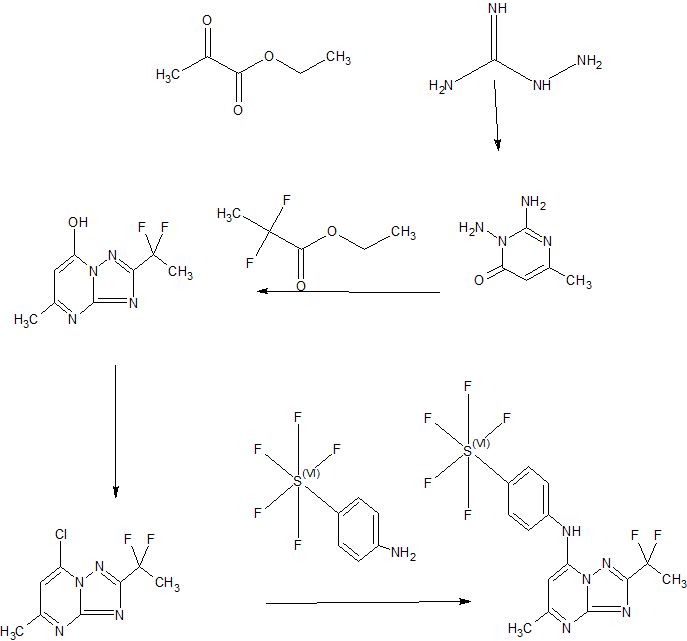
Example 44: Synthesis of 2-(l,l-difluoroethyl)-5-methyl-N-[4-(pentafluoro- 6– sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
A suspension of Intermediate 3 (5.84 g, 25.09 mmol) and 4-aminophenylsulfur pentafluoride (MANCHESTER, 5.5 g, 25.09 mmol) in ethanol (150 mL) was heated at 50 °C for 1 h. Heating resulted in the precipitation of a solid. The reaction mixture was concentrated under vacuum, redissolved in DCM (300 mL) and washed with aq. Na2C03 (2 x 350 mL). The organic layer was dried over Na2S04 and filtered. Then 8 g of silica gel were added and the mixture was concentrated under vacuum to dryness. The residue was purified (silica gel column, eluting with Hexane/EtOAc mixtures from 100:0 to 50:50%) to afford the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm: 10.60 (bs, 1H), 7.97 (d, 2H), 7.67 (d, 2H), 6.79 (s, 1H), 2.47 (s, 3H), 2.13 (t, 3H); [ES+ MS] m/z 416 (MH)+.
PAPER
Journal of Medicinal Chemistry (2011), 54(15), 5540-5561
http://pubs.acs.org/doi/abs/10.1021/jm200592f

Drug therapy is the mainstay of antimalarial therapy, yet current drugs are threatened by the development of resistance. In an effort to identify new potential antimalarials, we have undertaken a lead optimization program around our previously identified triazolopyrimidine-based series of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. The X-ray structure of PfDHODH was used to inform the medicinal chemistry program allowing the identification of a potent and selective inhibitor (DSM265) that acts through DHODH inhibition to kill both sensitive and drug resistant strains of the parasite. This compound has similar potency to chloroquine in the humanized SCID mouse P. falciparum model, can be synthesized by a simple route, and rodent pharmacokinetic studies demonstrated it has excellent oral bioavailability, a long half-life and low clearance. These studies have identified the first candidate in the triazolopyrimidine series to meet previously established progression criteria for efficacy and ADME properties, justifying further development of this compound toward clinical candidate statu
PAPER
Malaria persists as one of the most devastating global infectious diseases. The pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH) has been identified as a new malaria drug target, and a triazolopyrimidine-based DHODH inhibitor 1 (DSM265) is in clinical development. We sought to identify compounds with higher potency against PlasmodiumDHODH while showing greater selectivity toward animal DHODHs. Herein we describe a series of novel triazolopyrimidines wherein the p-SF5-aniline was replaced with substituted 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl amines. These compounds showed strong species selectivity, and several highly potent tetrahydro-2-naphthyl derivatives were identified. Compounds with halogen substitutions displayed sustained plasma levels after oral dosing in rodents leading to efficacy in the P. falciparum SCID mouse malaria model. These data suggest that tetrahydro-2-naphthyl derivatives have the potential to be efficacious for the treatment of malaria, but due to higher metabolic clearance than 1, they most likely would need to be part of a multidose regimen
Tetrahydro-2-naphthyl and 2-Indanyl Triazolopyrimidines TargetingPlasmodium falciparum Dihydroorotate Dehydrogenase Display Potent and Selective Antimalarial Activity
Sreekanth Kokkonda†, Xiaoyi Deng‡, Karen L. White§, Jose M. Coteron∥, Maria Marco∥, Laura de las Heras∥,John White†, Farah El Mazouni‡, Diana R. Tomchick⊥, Krishne Manjalanagara#, Kakali Rani Rudra#, Gong Chen§,Julia Morizzi§, Eileen Ryan§, Werner Kaminsky†, Didier Leroy∇, María Santos Martínez-Martínez∥, Maria Belen Jimenez-Diaz∥, Santiago Ferrer Bazaga∥, Iñigo Angulo-Barturen∥, David Waterson∇, Jeremy N. Burrows∇, Dave Matthews∇, Susan A. Charman§, Margaret A. Phillips*‡, and Pradipsinh K. Rathod*†
† Departments of Chemistry and Global Health, University of Washington, Seattle, Washington 98195, United States
‡Departments of Pharmacology and ⊥Biophysics, University of Texas Southwestern Medical Center at Dallas, 6001 Forest Park Blvd, Dallas, Texas 75390-9041, United States
§ Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
∥ GSK, Tres Cantos Medicines Development Campus, Severo Ochoa, Madrid 28760 Spain
# Syngene International Ltd., Bangalore 560 099, India
∇ Medicines for Malaria Venture, 1215 Geneva, Switzerland
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.6b00275
REFERENCES
1: Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El
Mazouni F, Lao Y, Kokkonda S, Tomchick DR, Deng X, Laird T, Bhatia SN, March S,
Ng CL, Fidock DA, Wittlin S, Lafuente-Monasterio M, Benito FJ, Alonso LM,
Martinez MS, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Haselden JN, Louttit
J, Cui Y, Sridhar A, Zeeman AM, Kocken C, Sauerwein R, Dechering K, Avery VM,
Duffy S, Delves M, Sinden R, Ruecker A, Wickham KS, Rochford R, Gahagen J, Iyer
L, Riccio E, Mirsalis J, Bathhurst I, Rueckle T, Ding X, Campo B, Leroy D, Rogers
MJ, Rathod PK, Burrows JN, Charman SA. A long-duration dihydroorotate
dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci
Transl Med. 2015 Jul 15;7(296):296ra111. doi: 10.1126/scitranslmed.aaa6645.
PubMed PMID: 26180101; PubMed Central PMCID: PMC4539048.
2: Held J, Jeyaraj S, Kreidenweiss A. Antimalarial compounds in Phase II clinical
development. Expert Opin Investig Drugs. 2015 Mar;24(3):363-82. doi:
10.1517/13543784.2015.1000483. Epub 2015 Jan 7. Review. PubMed PMID: 25563531.
3: Gamo FJ. Antimalarial drug resistance: new treatments options for Plasmodium.
Drug Discov Today Technol. 2014 Mar;11:81-88. doi: 10.1016/j.ddtec.2014.03.002.
Review. PubMed PMID: 24847657.
4: Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El
Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen
I, Ferrer SB, Jiménez-Díaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I,
Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA.
Structure-guided lead optimization of triazolopyrimidine-ring substituents
identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors
with clinical candidate potential. J Med Chem. 2011 Aug 11;54(15):5540-61. doi:
10.1021/jm200592f. Epub 2011 Jul 14. PubMed PMID: 21696174; PubMed Central PMCID:
PMC3156099.



/////DSM-265, PfSPZ, DSM-265, DSM 265, 1282041-94-4, (OC-6-21)-
FS(F)(F)(F)(C1=CC=C(NC2=CC(C)=NC3=NC(C(F)(F)C)=NN23)C=C1)F