












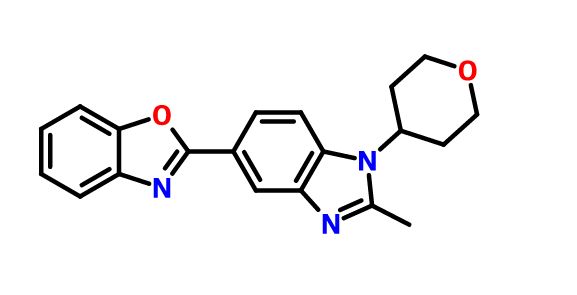
2-[2-Methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-1,3-benzoxazole Hemifumarate
Sumitomo Dainippon Pharma Company,
CAS FREE FORM 1256966-65-0
Benzoxazole, 2-[2-methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-
1H NMR (400 MHz, DMSO-d6)
13C NMR (100 MHz, DMSO-d6)


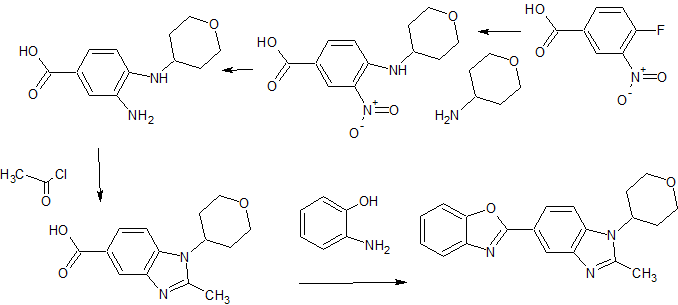
5- (benzoxazol-2-yl) -2-methyl -1-(tetrahydropyran-4-yl) benzimidazole eggplant flask (100 mL), 2- methyl-1- (tetrahydropyran – 4-yl) reference benzimidazole-5-carboxylic acid (example 4-3) (0.64 g, 2.46 mmol ), 2- amino-phenol (0.32 g, 2.95 mmol), and polyphosphoric acid (about 18 g) put, heated to 160 ℃, and the mixture was stirred for 17 hours. After cooling, ice was added, and the mixture was about pH 9 the liquid with concentrated aqueous ammonia (28%). Extraction with chloroform (about 50 mL X 3 times), dried over anhydrous magnesium sulfate, the crude product obtained by distilling off the solvent (0.08 g) PTLC (CHCl 3 by weight deploy purified), the title compound ( 0.002 g, 0.2% yield) was obtained as a yellow-brown semi-solid. 1H-NMR (CDCl 3 ) Deruta (Ppm): 1.88-1.92 (M, 2 H), 2.58-2.68 (M, 2 H), 2.70 (S, 3 H), 3.57-3.64 (M , 2 H), 4.21-4.25 (m , 2 H), 4.43-4.49 (m, 1 H), 7.29 (d, 1H, J = 9.2 Hz), 7.33-7.35 (m, 2 H ), 7.59-7.62 (m, 1 H ), 7.76-7.78 (m, 1 H), 8.18 (dd, 1 H, J = 8.6, 1.6 Hz), 8.57 (d, 1 H, J = 1.4 Hz).

PAPER

A short and practical synthetic route of a PDE4 inhibitor (1) was established by using Pd–Cu-catalyzed C–H/C–Br coupling of benzoxazole with a heteroaryl bromide. The combination of Pd(OAc)2-Cu(OTf)2-PPh3 was found to be effective for this key step. Furthermore, telescoping methods were adopted to improve the yield and manufacturing time, and a two-step synthesis of1 was accomplished in 71% overall yield.
Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide
///////////PDE4 inhibitor , Sumitomo Dainippon Pharma Company
Cc1nc3cc(ccc3n1C2CCOCC2)c4nc5ccccc5o4















