











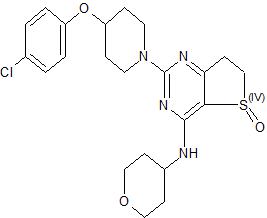
 R CONF SHOWN
R CONF SHOWN
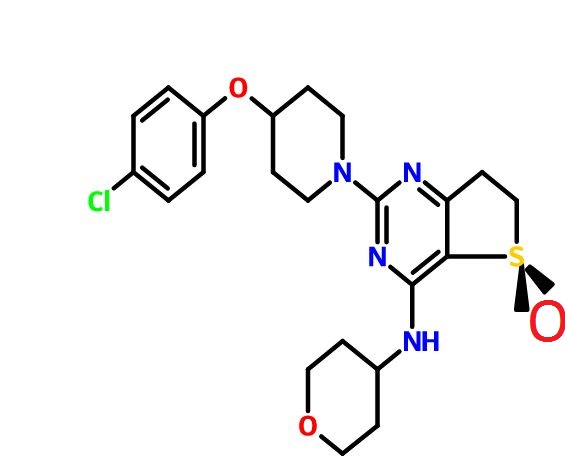
BI ?
(R)-2-(4-(4-Chlorophenoxy)piperidin-1-yl)-4-((tetrahydro-2H-pyran-4-yl)amino)-6,7-dihydrothieno[3,2-d]pyrimidine 5-Oxide
1H NMR (400 MHz, CDCl3) δ 1.49 (dq, J = 4.2, 11.8 Hz, 1H), 1.62 (dq, J = 4.2, 11.8 Hz, 1H), 1.74–1.89 (m, 3H), 1.90–2.02 (m, 3H), 2.96–3.07 (m, 2H), 3.29 (dt, J = 13.6, 8.4 Hz, 1H), 3.44 (ddd, J = 19.2, 11.2, 2.0 Hz, 2H), 3.62 (dt, J = 17.2, 7.8 Hz, 1H), 3.76 (m, 2H), 3.96 (dd, J = 15.6, 12.8 Hz, J = 2H), 4.09–3.99 (m, 3H), 4.51 (m, 1H), 6.21 (br d, J = 6.0 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H);
13C NMR (100 MHz, CDCl3) δ 30.4, 32.5, 32.7, 41.0, 47.2, 49.6, 66.9, 66.9, 72.9, 107.8, 117.5, 125.9, 129.5, 155.8, 158.9, 163.0, 174.6.
The use of phosphodiesterase type 4 (PDE4) inhibitors for the treatment of COPD (chronic obstructive pulmonary disease) by reducing inflammation and improving lung function is well documented. Given the potential therapeutic benefit offered by these compounds, a number of PDE4-selective inhibitors containing a dihydrothieno[3,2-d]pyrimidine core were identified as preclinical candidates in Boehringer Ingelheim Pharmaceuticals discovery laboratories

While the pathogenesis of chronic obstructive pulmonary disease (COPD) is incompletely understood, chronic inflammation is a major factor. In fact, the inflammatory response is abnormal, with CD8+ T-cells, CD68+ macrophages, and neutrophils predominating in the conducting airways, lung parenchyma, and pulmonary vasculature. Elevated levels of the second messenger cAMP can inhibit some inflammatory processes. Theophylline has long been used in treating asthma; it causes bronchodilation by inhibiting cyclic nucleotide phosphodiesterase (PDE), which inactivates cAMP. By inhibiting PDE, theophylline increases cAMP, inhibiting inflammation and relaxing airway smooth muscle. Rather than one PDE, there are now known to be more than 50, with differing activities, substrate preferences, and tissue distributions. Thus, the possibility exists of selectively inhibiting only the enzyme(s) in the tissue(s) of interest. PDE 4 is the primary cAMP-hydrolyzing enzyme in inflammatory and immune cells (macrophages, eosinophils, neutrophils). Inhibiting PDE 4 in these cells leads to increased cAMP levels, down-regulating the inflammatory response. Because PDE 4 is also expressed in airway smooth muscle and, in vitro, PDE 4 inhibitors relax lung smooth muscle, selective PDE 4 inhibitors are being developed for treating COPD. Clinical studies have been conducted with PDE 4 inhibitors;

Chronic obstructive pulmonary disease (COPD) is a serious and increasing global public health problem; physiologically, it is characterized by progressive, irreversible airflow obstruction and pathologically, by an abnormal airway inflammatory response to noxious particles or gases (MacNee 2005a). The COPD patient suffers a reduction in forced expiratory volume in 1 second (FEV1), a reduction in the ratio of FEV1 to forced vital capacity (FVC), compared with reference values, absolute reductions in expiratory airflow, and little improvement after treatment with an inhaled bronchodilator. Airflow limitation in COPD patients results from mucosal inflammation and edema, bronchoconstriction, increased secretions in the airways, and loss of elastic recoil. Patients with COPD can experience ‘exacerbations,’ involving rapid and prolonged worsening of symptoms (Seneff et al 1995; Connors et al 1996; Dewan et al 2000; Rodriguez-Roisin 2006; Mohan et al 2006). Many are idiopathic, though they often involve bacteria; airway inflammation in exacerbations can be caused or triggered by bacterial antigens (Murphy et al 2000; Blanchard 2002; Murphy 2006;Veeramachaneni and Sethi 2006). Increased IL-6, IL-1β, TNF-α, GRO-α, MCP-1, and IL-8 levels are found in COPD patient sputum; their levels increase further during exacerbations. COPD has many causes and significant differences in prognosis exist, depending on the cause (Barnes 1998; Madison and Irwin 1998).
COPD is already the fourth leading cause of death worldwide, according to the World Health Organization (WHO); the WHO estimates that by the year 2020, COPD will be the third-leading cause of death and the fifth-leading cause of disability worldwide (Murray and Lopez 1997). COPD is the fastest-growing cause of death in developed nations and is responsible for over 2.7 million deaths per year worldwide. In the US, there are currently estimated to be 16 million people with COPD. There are estimated to be up to 20 million sufferers in Japan, which has the world’s highest per capita cigarette consumption and a further 8–12 million in Europe. In 2000, COPD accounted for over 20 million outpatient visits, 3.4 million emergency room visits, 6 million hospitalizations, and 116,500 deaths in the US (National Center for Health Statistics 2002). Factors associated with COPD, including immobility, often lead to secondary health consequences (Polkey and Moxham 2006).
Risk factors for the development of COPD include cigarette smoking, and occupational exposure to dust and chemicals (Senior and Anthonisen 1998; Anthonisen et al 2002; Fabbri and Hurd 2003; Zaher et al 2004). Smoking is the most common cause of COPD and the underlying inflammation typically persists in ex-smokers. Oxidative stress from cigarette smoke is also an issue in COPD (Domej et al 2006). Despite this, relatively few smokers ever develop COPD (Siafakas and Tzortzaki 2002).
While many details of the pathogenesis of COPD remain unclear, chronic inflammation is now recognized as a major factor, predominantly in small airways and lung parenchyma, characterized by increased numbers of macrophages, neutrophils, and T-cells (Barnes 2000; Stockley 2002). As recently as 1995, the American Thoracic Society issued a statement defining COPD without mentioning the underlying inflammation (American Thoracic Society 1995). Since then, the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines have made it clear that chronic inflammation throughout the airways, parenchyma, and pulmonary vasculature plays a central role (Pauwels et al 2001; GOLD 2003). The comparatively recent realization of the role of airway inflammation in COPD has altered thinking with regard to potential therapies (Rogers and Giembycz 1998; Vignola 2004).
Most pharmacological therapies available for COPD, including bronchodilator and anti-inflammatory agents, were first developed for treating asthma. The mainstays of COPD treatment are inhaled corticosteroids (McEvoy and Niewoehner 1998; Borron and deBoisblanc 1998; Pauwels 2002; Gartlehner et al 2006;D’Souza 2006), supplemental oxygen (Petty 1998; Austin and Wood-Baker 2006), inhaled bronchodilators (Costello 1998; Doherty and Briggs 2004), and antibiotics (Taylor 1998), especially in severely affected patients (Anthonisen et al 1987; Saint et al 1995; Adams et al 2001; Miravitlles et al 2002; Donnelly and Rogers 2003; Sin et al 2003; Rabe 2006), though the use of antibiotics remains controversial (Ram et al 2006). Long-acting β2-agonists (LABAs) improve the mucociliary component of COPD. Combination therapy with LABAs and anticholinergic bronchodilators resulted in modest benefits and improved health-related quality of life (Buhl and Farmer 2005; Appleton et al 2006). Treatment with mucolytics reduced exacerbations and the number of days of disability (Poole and Black 2006). The combined use of inhaled corticosteroids and LABAs has been demonstrated to produce sustained improvements in FEV1 and positive effects on quality of life, number of hospitalizations, distance walked, and exacerbations (Mahler et al 2002;Szafranski et al 2003; Sin et al 2004; Miller-Larsson and Selroos 2006; van Schayck and Reid 2006). However, all of these treatments are essentially palliative and do not impact COPD progression (Hay 2000;Gamble et al 2003; Antoniu 2006a).
A further complication in drug development and therapy is that it can be difficult to determine the efficacy of therapy, because COPD has a long preclinical stage, is progressive, and patients generally do not present for treatment until their lung function is already seriously impaired. Moreover, because COPD involves irreversible loss of elasticity, destruction of the alveolar wall, and peribronchial fibrosis, there is often little room for clinical improvement.
Smoking cessation remains the most effective intervention for COPD. Indeed, to date, it is the only intervention shown to stop the decline in lung function, but it does not resolve the underlying inflammation, which persists even in ex-smokers. Smoking cessation is typically best achieved by a multifactor approach, including the use of bupropion, a nicotine replacement product, and behavior modification (Richmond and Zwar 2003).
In COPD, there is an abnormal inflammatory response, characterized by a predominance of CD8+ T-cells, CD68+ macrophages, and neutrophils in the conducting airways, lung parenchyma, and pulmonary vasculature (Soto and Hanania 2005; O’Donnell et al 2006; Wright and Churg 2006). Inflammatory mediators involved in COPD include lipids, inflammatory peptides, reactive oxygen and nitrogen species, chemokines, cytokines, and growth factors. COPD pathology also includes airway remodeling and mucociliary dysfunction (mucus hypersecretion and decreased mucus transport). Corticosteroids reduce the number of mast cells, but CD8+ and CD68+ cells, and neutrophils, are little affected (Jeffery 2005). Inflammation in COPD is not suppressed by corticosteroids, consistent with it being neutrophil-, not eosinophil-mediated. Corticosteroids also do not inhibit the increased concentrations of IL-8 and TNF-α (both neutrophil chemoattractants) found in induced sputum from COPD patients. Neutrophil-derived proteases, including neutrophil elastase and matrix metalloproteinases (MMPs), are involved in the inflammatory process and are responsible for the destruction of elastin fibers in the lung parenchyma (Mercer et al 2005; Gueders et al 2006). MMPs play important roles in the proteolytic degradation of extracellular matrix (ECM), in physiological and pathological processes (Corbel, Belleguic et al 2002). PDE 4 inhibitors can reduce MMP activity and the production of MMPs in human lung fibroblasts stimulated with pro-inflammatory cytokines (Lagente et al 2005). In COPD, abnormal remodeling results in increased deposition of ECM and collagen in lungs, because of an imbalance of MMPs and TIMPs (Jeffery 2001). Fibroblast/myofibroblast proliferation and activation also occur, increasing production of ECM-degrading enzymes (Crouch 1990; Segura-Valdez et al 2000). Additionally, over-expression of cytokines and growth factors stimulates lung fibroblasts to synthesize increased amounts of collagen and MMPs, including MMP-1 (collagenase-1) and MMP-2 and MMP-9 (gelatinases A and B) (Sasaki et al 2000; Zhu et al 2001).
It is now generally accepted that bronchial asthma is also a chronic inflammatory disease (Barnes et al 1988;Barnes 1995). The central role of inflammation of the airways in asthma’s pathogenesis is consistent with the efficacy of corticosteroids in controlling clinical symptoms. Eosinophils are important in initiating and continuing the inflammatory state (Holgate et al 1987; Bruijnzeel 1989; Underwood et al 1994; Teixeira et al 1997), while other inflammatory cells, including lymphocytes, also infiltrate the airways (Holgate et al 1987;Teixeira et al 1997). The familiar acute symptoms of asthma are the result of airway smooth muscle contraction. While recognition of the key role of inflammation has led to an emphasis on anti-inflammatory therapy in asthma, a significant minority of patients remains poorly controlled and some exhibit accelerated declines in lung function, consistent with airway remodeling (Martin and Reid 2006). Reversal or prevention of structural changes in remodeling may require additional therapy (Burgess et al 2006).
There is currently no cure for asthma; treatment depends primarily on inhaled glucocorticoids to reduce inflammation (Taylor 1998; Petty 1998), and inhaled bronchodilators to reduce symptoms (Torphy 1994;Costello 1998; Georgitis 1999; DeKorte 2003). Such treatments, however, do not address disease progression.
COPD and asthma are both characterized by airflow obstruction, but they are distinct in terms of risk factors and clinical presentation. While both involve chronic inflammation and cellular infiltration and activation, different cell types are implicated and there are differences in the inflammatory states (Giembycz 2000;Fabbri and Hurd 2003; Barnes 2006). In COPD, neutrophil infiltration into the airways and their activation appear to be key (Stockley 2002); in asthma, the inflammatory response involves airway infiltration by activated eosinophils and lymphocytes, and T-cell activation of the allergic response (Holgate et al 1987;Saetta et al 1998; Barnes 2006). While macrophages are present in both conditions, the major controller cells are CD8+ T-cells in COPD (O’Shaughnessy et al 1997; Saetta et al 1998) and CD4+ T-cells in asthma. IL-1, IL-8, and TNF-α are the key cytokines in COPD, while in asthma, IL-4, IL-5, and IL-13 are more important. There are differences in histopathological features of lung biopsies between COPD patients and asthmatics; COPD patients have many fewer eosinophils in lung tissue than asthmatics.
While the early phases of COPD and asthma are distinguishable, there are common features, including airway hyper-responsiveness and mucus hypersecretion. MUC5AC is a major mucin gene expressed in the airways; its expression is increased in COPD and asthmatic patients. At least in vitro, epidermal growth factor stimulates MUC5AC mRNA and protein expression; this can be reversed by PDE 4 inhibitors, which may contribute to their clinical efficacy in COPD and asthma (Mata et al 2005). Similar structural and fibrotic changes make COPD and asthma much less distinguishable in extreme cases; the chronic phases of both involve inflammatory responses, alveolar detachment, mucus hypersecretion, and subepithelial fibrosis. The two conditions have been linked epidemiologically; adults with asthma are up to 12 times more likely to develop COPD over time than those without (Guerra 2005).


PAPER

A practical, safe, and efficient process for the synthesis of PDE4 (phosphodiesterase type 4) inhibitors represented by 1 and 2 was developed and demonstrated on a multi-kilogram scale. Key aspects of the process include the regioselective synthesis of dihydrothieno[3,2-d]pyrimidine-2,4-diol 9 and the asymmetric sulfur oxidation of intermediate 11.
Development of a Practical Process for the Synthesis of PDE4 Inhibitors
PDE 4 in COPD
With regard to COPD, PDE 4 is the primary cAMP-hydrolyzing enzyme in inflammatory and immune cells, especially macrophages, eosinophils, and neutrophils, all of which are found in the lungs of COPD and asthma patients (Torphy et al 1992; Karlsson and Aldous 1997; De Brito et al 1997; Wang et al 1999;Torphy and Page 2000). Inhibition of PDE 4 leads to elevated cAMP levels in these cells, down-regulating the inflammatory response (Dyke and Montana 2002).
PDE 4 has also attracted much attention because it is expressed in airway smooth muscle (Ashton et al 1994;Undem et al 1994; Nicholson et al 1995; Kerstjens and Timens 2003; Mehats et al 2003; Lipworth 2005; Fan Chung 2006). In vitro, PDE 4 inhibitors relax lung smooth muscle (Undem et al 1994; Dent and Giembycz 1995). In COPD and asthma, a selective PDE 4 inhibitor with combined bronchodilatory and anti-inflammatory properties would seem desirable (Nicholson and Shahid 1994; Lombardo 1995; Palfreyman 1995; Cavalia and Frith 1995; Palfreyman and Souness 1996; Karlsson and Aldous 1997; Compton et al 2001; Giembycz 2002; Jacob et al 2002; Soto and Hanania 2005).
PDE 4 inhibitors in COPD
So, because PDE 4 inhibitors suppress inflammatory functions in several cell types involved in COPD and asthma (Huang and Mancini 2006) and because, at least in vitro, PDE 4 inhibitors relax lung smooth muscle, selective PDE 4 inhibitors, originally intended for use in treating depression (Renau 2004), have been developed for the treatment of COPD and asthma (Torphy et al 1999; Spina 2000; Huang et al 2001; Spina 2004; Giembycz 2005a, 2005b; Lagente et al 2005; Boswell-Smith, Spina et al 2006). PDE 4 enzymes are strongly inhibited by the antidepressant drug rolipram (Pinto et al 1993), which decreases the influx of inflammatory cells at sites of inflammation (Lagente et al 1994; Lagente et al 1995; Alves et al 1996). PDE 4 inhibitors down-regulate cytokine production in inflammatory cells, in vivo and in vitro (Undem et al 1994;Dent and Giembycz 1995). TNF-α is an important inflammatory cytokine in COPD; its release is reduced by PDE 4 inhibitors (Souness et al 1996; Chambers et al 1997; Griswold et al 1998; Gonçalves de Moraes et al 1998; Corbel, Belleguic et al 2002). Some PDE 4 inhibitors, including cilomilast and AWD 12-281, can inhibit neutrophil degranulation, a property not shared by theophylline (Ezeamuzie 2001; Jones et al 2005). PDE 4 inhibitors reduce overproduction of other pro-inflammatory mediators, including arachidonic acid and leukotrienes (Torphy 1998). PDE 4 inhibitors also inhibit cellular trafficking and microvascular leakage, production of reactive oxygen species, and cell adhesion molecule expression in vitro and in vivo (Sanz et al 2005). PDE 4 inhibitors, including cilomilast and CI-1044, inhibit LPS-stimulated TNF-α production in whole blood from COPD patients (Burnouf et al 2000; Ouagued et al 2005).
There are now thought to be at least four PDE 4s, A, B, C, and D, derived from four genes (Lobbam et al 1994; Muller et al 1996; Torphy 1998; Conti and Jin 1999; Matsumoto et al 2003). Alternative splicing and alternative promoters add further complexity (Manganiello et al 1995; Horton et al 1995; Torphy 1998). Indeed, the four genes encode more than 16 PDE 4 isoforms, which can be divided into short (∼65–75 kDa) and long forms (∼80–130 kDa); the difference between the short and long forms lies in the N-terminal region (Bolger et al 1997; Huston et al 2006). PDE 4 isoforms are regulated by extracellular signal-related protein kinase (ERK), which can phosphorylate PDE 4 (Houslay and Adams 2003).
The four PDE 4 genes are differentially expressed in various tissues (Silver et al 1988; Lobbam et al 1994;Manganiello et al 1995; Horton et al 1995; Muller et al 1996; Torphy 1998). PDE 4A is expressed in many tissues, but not in neutrophils (Wang et al 1999). PDE 4B is also widely expressed and is the predominant PDE 4 subtype in monocytes and neutrophils (Wang et al 1999), but is not found in cortex or epithelial cells (Jin et al 1998). Upregulation of the PDE 4B enzyme in response to pro-inflammatory agents suggest that it has a role in inflammatory processes (Manning et al 1999). PDE 4C is expressed in lung and testis, but not in circulating inflammatory cells, cortex, or hippocampus (Obernolte et al 1997; Manning et al 1999; Martin-Chouly et al 2004). PDE 4D is highly expressed in lung, cortex, cerebellum, and T-cells (Erdogan and Houslay 1997; Jin et al 1998). PDE 4D also plays an important role in airway smooth muscle contraction (Mehats et al 2003).
A major issue with early PDE 4 inhibitors was their side effect profile; the signature side effects are largely gastrointestinal (nausea, vomiting, increased gastric acid secretion) and limited the therapeutic use of PDE 4 inhibitors (Dyke and Montana 2002). The second generation of more selective inhibitors, such as cilomilast and roflumilast, have improved side effect profiles and have shown clinical efficacy in COPD and asthma (Barnette 1999; Spina 2000; Lagente et al 2005). However, even cilomilast and roflumilast, the most advanced clinical candidates, discussed below, cause some degree of emesis (Spina 2003).
It is now thought that the desirable anti-inflammatory properties and unwanted side effects of nausea and emesis are associated with distinct biochemical activities (Torphy et al 1992; Jacobitz et al 1996; Barnette et al 1996; Souness et al 1997; Souness and Rao 1997). Specifically, the side effects are believed to be associated with the so-called ‘high-affinity rolipram binding site’ (HARBS) (Barnette et al 1995; Muller et al 1996; Jacobitz et al 1996; Kelly et al 1996; Torphy 1998) and/or inhibition of the form of PDE 4 found in the CNS (Barnette et al 1996). The exact nature of HARBS remains unclear, although it has been described as a conformer of PDE 4 (Souness and Rao 1997; Barnette et al 1998). Using mice deficient in PDE 4B or PDE 4D, it appears that emesis is the result of selective inhibition of PDE 4D (Robichaud et al 2002; Lipworth 2005), which is unfortunate, because the most clinically advanced PDE 4 inhibitors are selective for PDE 4D. Also, from animal studies, it appears that the nausea and vomiting are produced via the CNS, though there may also be direct effects on the gastrointestinal system (Barnette 1999).
While beyond the scope of this review, it has been proposed that PDE 4 inhibitors may be useful in treating inflammatory bowel disease (Banner and Trevethick 2004), cystic fibrosis (Liu et al 2005), pulmonary arterial hypertension (Growcott et al 2006), myeloid and lymphoid malignancies (Lerner and Epstein 2006), Alzheimer’s disease (Ghavami et al 2006), rheumatoid arthritis and multiple sclerosis (Dyke and Montana 2002), infection-induced preterm labor (Oger et al 2004), depression (Wong et al 2006), and allergic disease (Crocker and Townley 1999). Varying degrees of in vitro, in vivo, and clinical data exist to support these claims.
So, after that theoretical buildup, we reach the proof of the pudding; clinical studies have been conducted with PDE 4 inhibitors. A potent, but not-very-selective, PDE 4 inhibitor is approved in Japan and is used clinically, including for treating asthma. Another is awaiting approval in the US. One is in advanced clinical development and others are at earlier stages.
REF
Pouzet, P.; Hoenke, C.; Martyres, D.; Nickolaus, P.; Jung, B.; Hamman, H. Dihydrothienopyrimidines for the treatment of inflammatory diseases. PatentWO 2006111549 A1, October 26, 2006.
Ohnacker, G.; Woitun, E. Novel dihydrothieno[3, 2-d]pyrimidines. U.S. Patent US 3,318,881, May 9, 1967.
/////PDE4 Inhibitors, Boehringer Ingelheim Pharmaceuticals, BI ?, PRECLINICAL, 1910076-27-5
Clc1ccc(cc1)OC2CCN(CC2)c4nc(NC3CCOCC3)c5c(n4)CCS5=O

Sorry, the comment form is closed at this time.