 .
.
Optimisation and real time reaction monitoring of the synthesis of 2-fluoromalonate esters by direct fluorination using fluorine gas is reported. An assessment of green metrics including atom economy and process mass intensity factors, demonstrates that the one-step selective direct fluorination process compares very favourably with established multistep processes for the synthesis of fluoromalonates.
 .
.
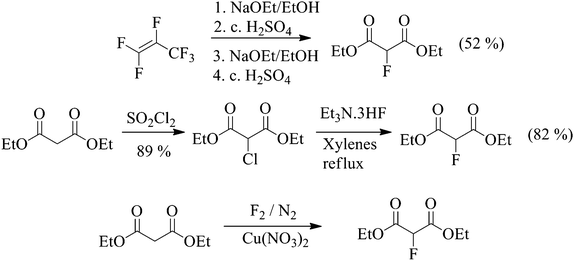
Scheme 2 Synthetic routes to 2-fluoromalonate esters.
There are three realistic, low-cost synthetic strategies available for the large scale manufacture of diethyl 2-fluoromalonate ester (Scheme 2) which involve reaction of ethanol with hexafluoropropene (HFP), halogen exchange (Halex)and selective direct fluorination processes. Other syntheses of fluoromalonate esters using electrophilic fluorinating agents such as Selectfluor™ are possible, but are not sufficiently commercially attractive to be considered for manufacture on the large scale.
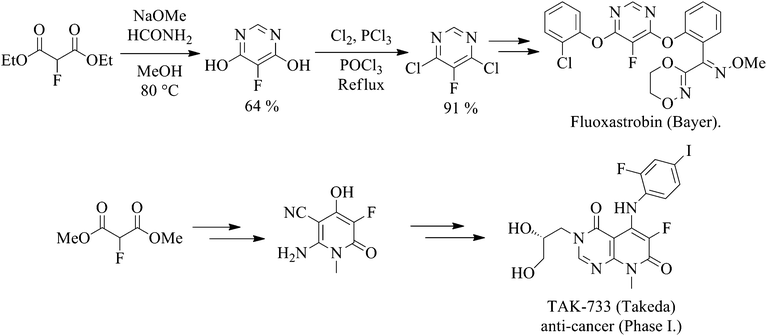
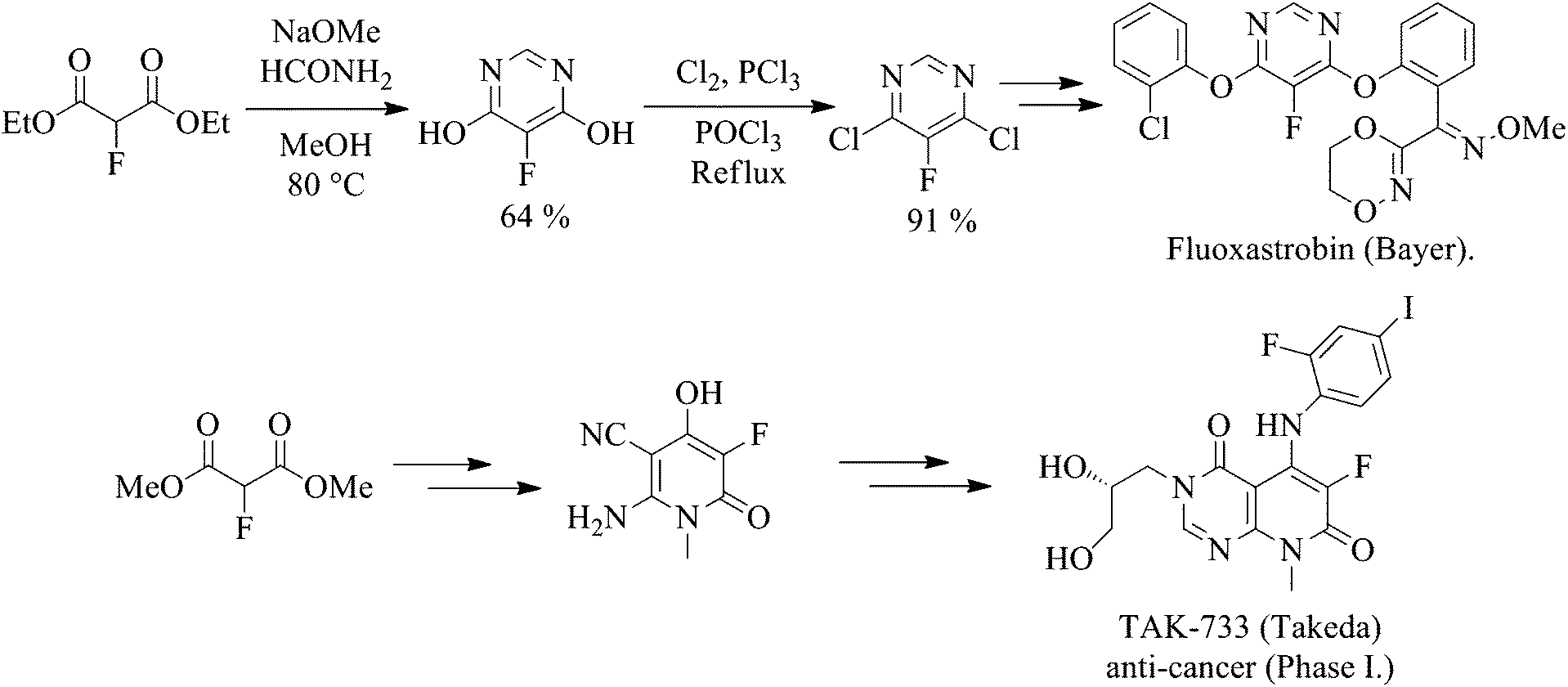
A growing number of patents utilising fluoromalonate as a substrate for the synthesis of a range of biologically active systems have been published For example, Fluoxastrobin (Fandango®), a fungicide marketed by Bayer CropScience that has achieved global annual sales of over €140 m since its launch in 2005, and TAK-733, an anti-cancer drug candidate, employ 2-fluoromalonate esters as the key fluorinated starting material (Scheme 1).
 |
|
Scheme 1 2-Fluoromalonate esters used in the synthesis of Fluoxastrobin and TAK-733. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Before a comparison of the green metrics between the three possible, economically viable large scale processes for the synthesis of fluoromalonate esters (Scheme 2) could be carried out, some primary goals for the optimisation of the process were targeted: complete conversion of the starting material is essential because it can be difficult to separate the starting material from the desired monofluorinated product by simple distillation; fluorine gas usage should be minimised because neutralisation of excess reagent could potentially generate significant amounts of waste; reduction in volumes of solvents used to reduce waste streams and overall intensification of the fluorination process and replacement and/or reduction of all environmentally harmful solvents used.
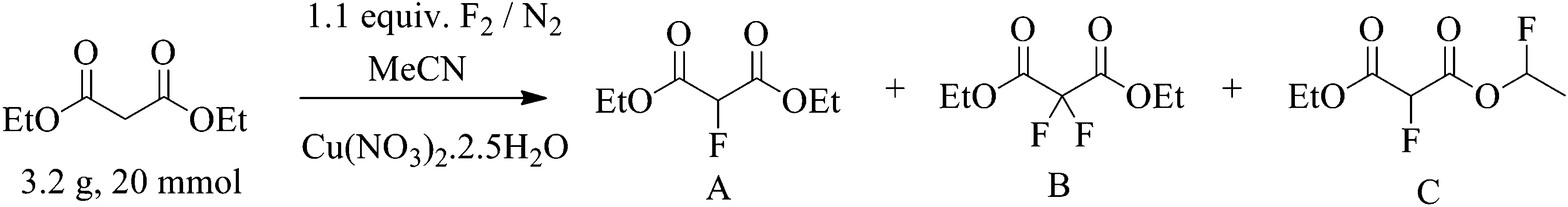
Conventional batch direct fluorination reactions of malonate esters were carried out in glassware vessels by introduction of fluorine gas, as a 10% or 20% mixture in nitrogen (v/v), at a prescribed rate via a gas mass flow controller into a solution of malonate ester and copper nitrate catalyst in acetonitrile using equipment described previously.
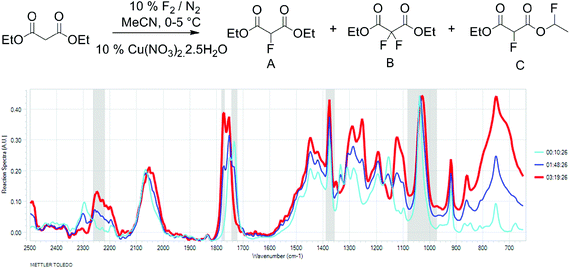
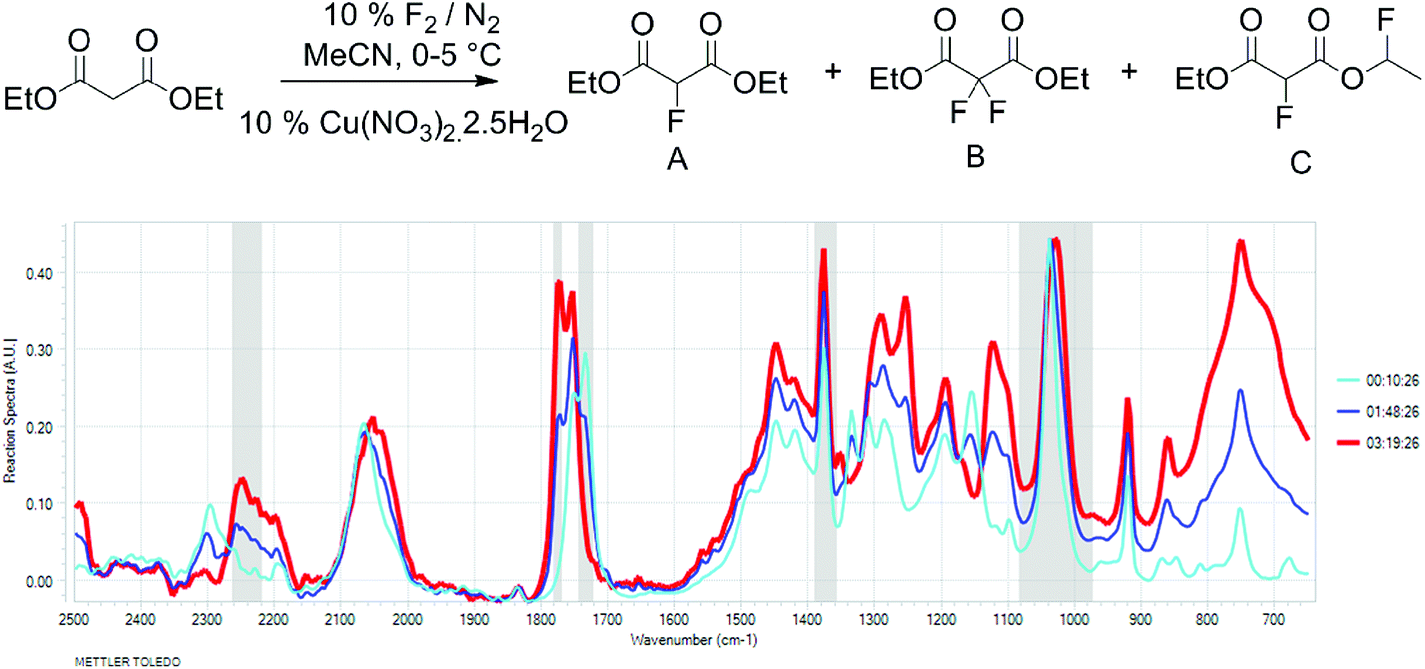
To better understand the relationship between fluorine gas introduction and rate of conversion, real time IR spectroscopic monitoring of the reaction was chosen as the most suitable technique. The use of the ReactIR technique was enabled by a sufficient difference in the carbonyl group stretching frequencies (1734 cm−1 for diethyl malonate and 1775 cm−1 for diethyl 2-fluoromalonate) and provided an in situ reaction profile (Fig. 1).
 |
|
Fig. 1 IR spectra of the fluorination reaction at 0% (light blue), 50% (dark blue) and 100% (red) conversions. |
|
|
|
|
|
|
|
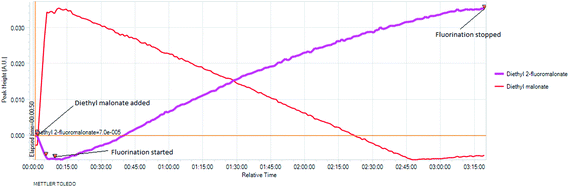
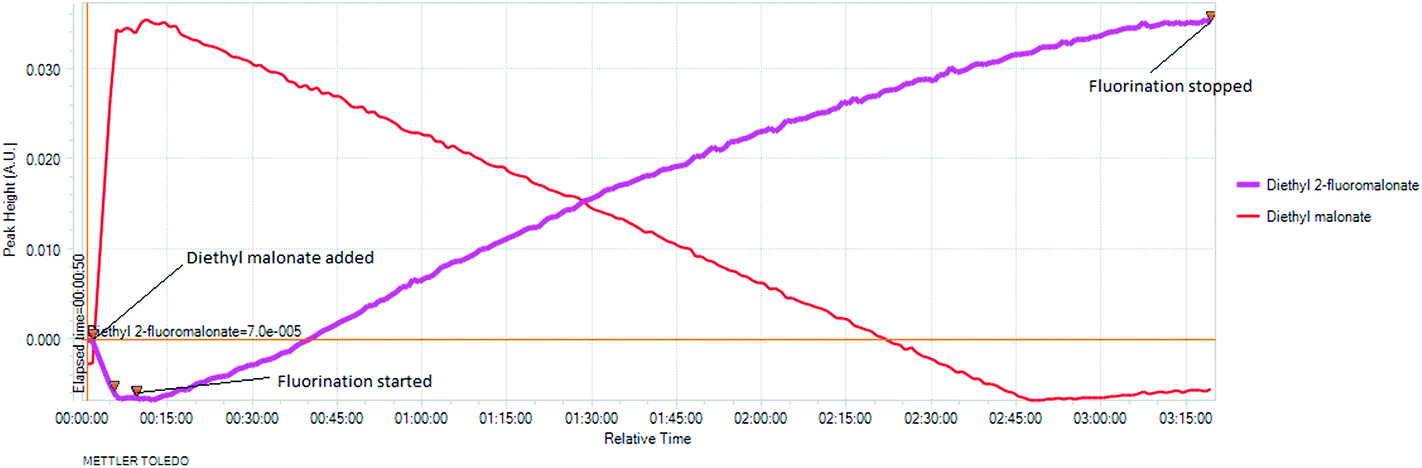
The real time reaction monitoring (Fig. 1 and 2) revealed that the reaction begins instantly upon initiation of fluorine introduction and the reaction conversion is directly proportional to the amount of fluorine gas passed into the reaction vessel. When the intensity of the fluoromalonate carbonyl peak (1775 cm−1) reached a maximum, the introduction of fluorine gas was stopped and the crude reaction mixture was analysed by 1H and 19F NMR spectroscopy. Complete conversion of the starting material was observed and diethyl fluoromalonate was formed with 93% selectivity after introducing 1.1 equivalents of fluorine into the reaction mixture. The small excess of fluorine explains the unexpectedly small amount of difluorinated side products B and C (4.5 and 2.5% respectively) which were the major impurities (6.5 and 9% respectively) when larger excess of fluorine gas (1.8 eq.) was used.
 |
|
Fig. 2 In situ monitoring of the fluorination of diethyl malonate. |
|
|
|
|
|
The effect of concentration of fluorine in nitrogen, reaction temperature, copper nitrate catalyst loading and concentration of malonate substrate in acetonitrile were varied to optimise the fluorination process (Table 1). Additionally, reactions described in Table 1 allowed an assessment of various factors that have a major influence on the environmental impact of the process such as solvent usage, reaction temperature and the amount and composition of waste generated. In each case 20 mmol (3.20 g) of diethyl malonate was used as substrate and the isolated mass balance of crude material obtained after work-up was recorded along with the conversion of starting material and yield of fluorinated products (Table 1).
Table 1 Fluorination of diethyl malonate ester using fluorine gas catalysed by Cu(NO3)2·2.5H2O
|
|
| Entry no. |
T/°C |
C malonate (mol L−1) |
Catalyst (mol%) |
F 2 in N2 (% v/v) |
Conversion (1H NMR) |
A/B/C ratio (19F NMR) |
Isolated weight |
| 1 |
0–5 |
1.0 |
10 |
10 |
100% |
93.5/4.5/2 |
3.37 g |
| 2 |
0–5 |
1.5 |
10 |
10 |
100% |
94/4/2 |
3.30 g |
| 3 |
0–5 |
1.0 |
5 |
10 |
97% |
95/4/1 |
3.53 g |
| 4 |
0–5 |
1.0 |
2.5 |
10 |
82% |
95/4/1 |
3.51 g |
| 5 |
RT |
1.0 |
10 |
10 |
56% |
97.5/1.5/1 |
3.33 g |
| 6 |
0–5 |
1.0 |
10 |
15 |
85% |
97.5/1.5/1 |
3.47 g |
| 7 |
0–5 |
1.0 |
10 |
20 |
100% |
94/3/3 |
3.50 g |
| 8 |
0–5 |
2.0 |
5 |
20 |
52% |
92/5/3 |
3.40 g |
In all cases, small quantities of side products were formed which were identified by 19F NMR and these originate from two different processes: 3,3-difluoromalonate is produced from enolisation of diethyl fluoromalonate which is much slower than enolisation of the diethyl malonate substrate, while the fluoroethyl fluoromalonate is postulated to form via an electrophilic process.
The data in Table 1 suggest that the concentration of the malonate ester substrate in acetonitrile has no apparent effect on the outcome of the reaction although solvent is required for these reactions because diethyl malonate does not dissolve the catalyst. Additionally, the use of high dielectric constant media, such as acetonitrile, have been found to be beneficial for the control of selectivity of electrophilic direct fluorination processes. For convenience, a 1.5 M concentration of malonate in acetonitrile was chosen as the optimal conditions which is approximately 5 mL solvent per 1 mL of diethyl malonate.
The concentration of fluorine gas, between 10–20% v/v in nitrogen, does not affect the selectivity of the reaction and the quality of the product either, as exemplified by the product mixtures obtained from reactions 1, 2 and 7 which have identical compositions. In contrast, carrying out fluorination reactions at room temperature rather than cooling the reaction mixture to 0–5 °C leads to increased catalyst decomposition which results in an insoluble copper species that on occasion blocked the fluorine gas inlet tube. In addition, without cooling, the exothermic nature of this fluorination reaction led to a slight reaction temperature increase (from 20 to 29 °C in a small scale laboratory experiment) resulting in loss of some solvent and some decomposition of the catalyst and product degradation.
Lowering the concentration of the copper nitrate catalyst led to a significantly slower reaction as would be expected and required the use of a larger excess of fluorine gas to enable sufficiently high conversion. For example, the reaction proceeded in the presence of only 2.5 mol% catalyst, but in this case 40% excess fluorine was required to reach 100% conversion.
Typical literature work-up procedures for direct fluorination reactions involve pouring the reaction mixture into 3 to 5 volumes of water and extracting the resulting mixture three times with dichloromethane. The combined organic fraction is typically washed with water, saturated sodium bicarbonate solution and dried over sodium sulfate before evaporation of the solvent to give the crude reaction product. We sought to improve the work-up to enable recycling of the reaction solvent and substitute the use of environmentally harmful dichloromethane in the reaction work-up stage. Upon completion of fluorine gas addition, acetonitrile was evaporated for reuse and then the residue was partitioned between ethyl acetate and water, the organic phase was washed with water, saturated Na2CO3 solution and saturated brine and dried prior to evaporation under reduced pressure. Modification of the workup procedure in this manner enables the recovery of acetonitrile and ethyl acetate and significantly reduces the amount of aqueous waste generated. When direct reuse of the recovered acetonitrile was attempted, a copper containing precipitate was formed presumably because of the high HF content of the solvent (0.63 M by titration). Therefore, before reuse of the solvent, HF must be removed. Stirring the recovered reaction solvent with solid Na2CO3 lowered the acid content to an acceptable level (0.04 M) and when a second fluorination reaction was carried out in the recovered, neutralised acetonitrile, no change in the fluorination reaction profile was observed.
Upon completion of these optimisation studies, selective fluorination reactions of malonate esters were scaled up to 40 g scale in the laboratory without experiencing any change in product profile. Isolation of significant quantities of monofluoromalonate A crude product (99% yield, 95% purity) was achieved which could be used in the subsequent cyclisation processes described below without further purification or, if high purity material was required, could be purified by fractional vacuum distillation (bp. 102–103 °C, 18 mbar) to produce 99% pure material in 77% yield.
Related malonate esters were also subjected to direct fluorination using the optimised conditions established above. In the case of di-tert-butyl malonate, fluorination was carried out on 12 g scale. 100% conversion was reached after the introduction of 1.2 equivalents of fluorine gas and the desired product was isolated in 96% yield. The purity of the crude product was higher than 97% by 1H and 19F NMR spectroscopy without any further purification and as expected, the only side product was the 2,2-difluorinated product (Scheme 3).
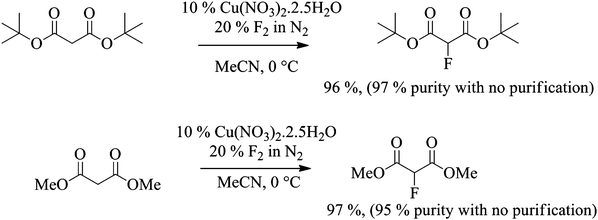 |
|
Scheme 3 Fluorination of di-methyl and di-tert-butyl malonates. |
|
Diethyl fluoromalonate large scale fluorination
Diethyl malonate (40.0 g, 0.25 mol) and copper nitrate hydrate (Cu(NO3)2·2.5H2O; 5.81 g, 25 mmol) were dissolved in acetonitrile (200 mL) and placed in 500 mL fluorination vessel, cooled to 0–5 °C and stirred at 650 rpm using an overhead stirrer. After purging the system with N2 for 5 minutes, fluorine gas (20% v/v in N2, 80 mL min−1, 265 mmol) was introduced into the mixture for 6 hours and 30 minutes. The reactor was purged with nitrogen for 10 minutes, the solvent removed in vacuo and the residue partitioned between water (50 mL) and ethyl acetate (50 mL). The aqueous phase was extracted once more with ethyl acetate (50 mL) and the combined organic layers were washed with saturated NaHCO3 (25 mL) and brine (20 mL). After drying over sodium sulfate, the solvent was evaporated to leave diethyl 2-fluoromalonate (44.4 g, 99% yield, 95% purity) as a light yellow, transparent liquid. This crude product was distilled to afford high purity fluoromalonate (34.7 g, 77% yield, 99%+ purity) as a colourless liquid, bp. 102–103 °C (18 mbar), (lit.: 110–112 °C, 29 mbar), spectroscopic data as above………N. Ishikawa, A. Takaoka and M. K. Ibrahim, J. Fluorine Chem., 1984, 25, 203–212 CrossRef CAS.
PAPER
REF
DOI:
10.1039/C5GC00402K (Paper)
Green Chem., 2015,
17, 3000-3009
Fluorine gas for life science syntheses: green metrics to assess selective direct fluorination for the synthesis of 2-fluoromalonate esters†
Received 19th February 2015 , Accepted 17th March 2015
First published on the web 17th March 2015
Optimisation and real time reaction monitoring of the synthesis of 2-fluoromalonate esters by direct fluorination using fluorine gas is reported. An assessment of green metrics including atom economy and process mass intensity factors, demonstrates that the one-step selective direct fluorination process compares very favourably with established multistep processes for the synthesis of fluoromalonates.
Paper
Fluorine gas for life science syntheses: green metrics to assess selective direct fluorination for the synthesis of 2-fluoromalonate esters
Green Chem., 2015,17, 3000-3009
DOI: 10.1039/C5GC00402K












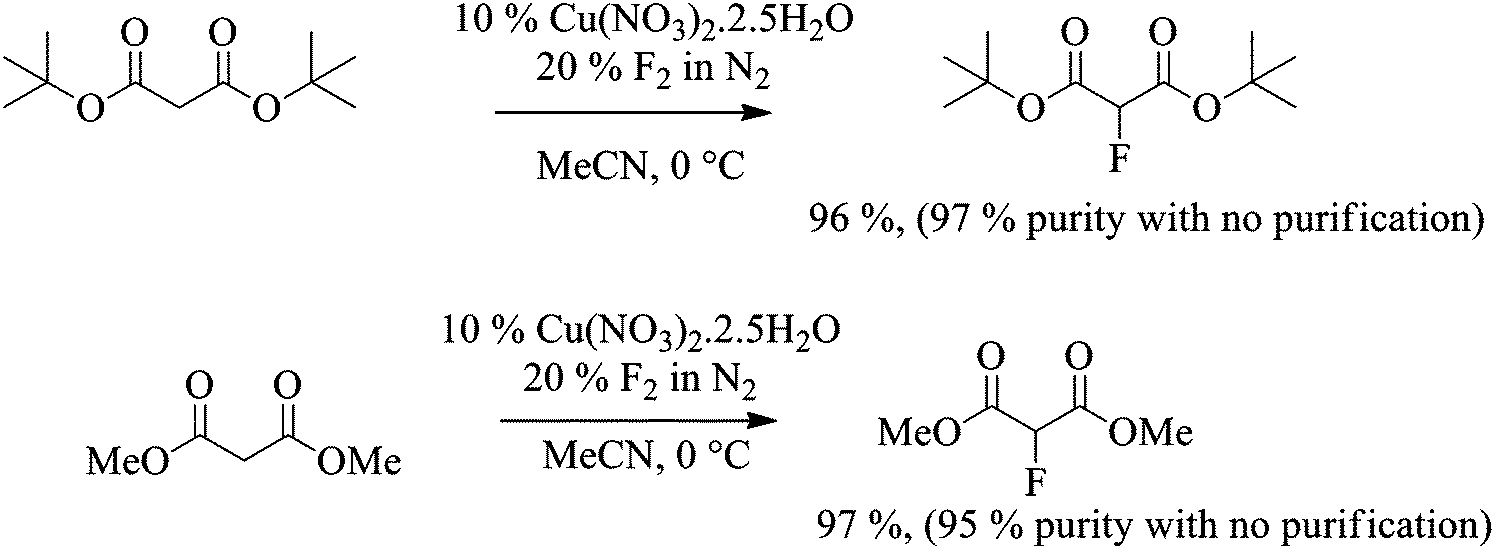

Sorry, the comment form is closed at this time.