












Telescoping multistep reactions
The synthesis of fine chemicals sometimes requires multiple reactions and tedious work-up between each step is often necessary. Purification may involve the addition of a quenching reagent, multiple aqueous and organic extractions, the addition of a drying agent, filtration, evaporation, and further purification by chromatography, distillation, or recrystallization. These operations all require significant input of energy and materials that ultimately end up as large amounts of waste. Methods and technologies that eliminate or simplify one or many of these steps can make a significant influence on the environmental impact of a multistep chemical synthesis. Continuous processing is particularly suitable for ‘telescoping’ reaction sequences, and many methods have been developed to facilitate this.1
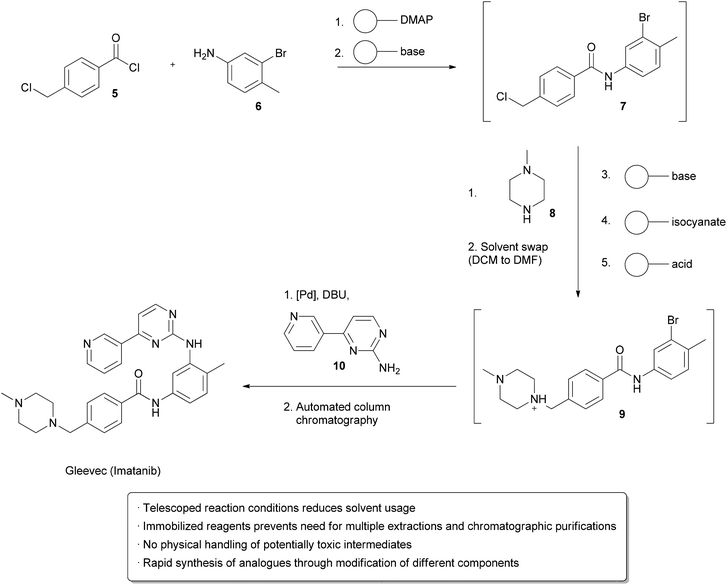
One strategy utilizes solid supported reagents packed into columns which allow starting materials to flow in and product to be collected at the outlet without requiring separation of the spent reagent. Different columns may be linked in series, allowing multistep processes to take place. Extra operations may also be necessary, such as solvent changes or the removal of unwanted side products. Methods for automating these processes have also been developed. An example from the Ley group illustrates many of these technologies in the design of a single apparatus to continuously prepareImatinib (Gleevec) from simple starting materials (Scheme 1).2Acid chloride 5 and aniline 6 in DCM were flowed through a cartridge containing immobilized DMAP as a nucleophilic catalyst, followed by a basic cartridge to scavenge any remaining 5. The formation of the amide 7 was monitored by an in-line UV spectrometer and subsequently added to a vial containing piperazine 8 in DMF at 50 °C, which facilitated evaporation of the DCM. Once a particular amount of 7 was obtained, as indicated by the UV spectrometer, a connected autosampler would collect this solution and pump it through an immobilized base to induce a substitution reaction, followed by an immobilized isonitrile to scavenge any remaining 8. An immobilized acid was used to ‘catch’ amine 9 through protonation, allowing unreacted 7 to go to waste. ‘Release’ of 9 through deprotonation followed by the addition of aniline 10 and a palladium catalyst facilitated a cross-coupling reaction, furnishing the crude Imatinib, which was then evaporated onto a silica gel column for automated chromatography. Pure product was isolated in 32% overall yield and >95% purity. While not explicitly demonstrated, the possibility of using this apparatus to form analogs by using modified starting materials is proposed. The ability to perform multi-step synthesis of pharmaceuticals without handling of the intermediates is particularly interesting, as exposure to these species can be hazardous.
 |
||
| Scheme 1 Multistep synthesis of Imatinib (Gleevec). |
||
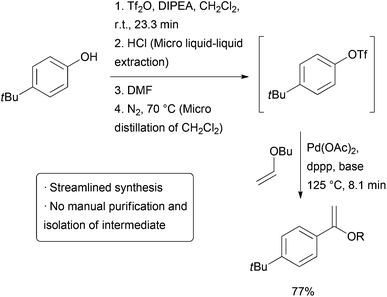
The above example utilizes packed cartridges of scavengers to effect purification. An alternative method is to more closely emulate typical batch purification operations such as distillation andextraction, but on a small, continuous scale. Several different ‘chip’ purification devices have been developed for this purpose.3-12 Some of these technologies were used together in a combined triflation/Heck reaction of phenols (Scheme2). After the initial triflation step in dichloromethane, the product is combined with a stream of aqueous HCl and passed on to a chip containing a membrane that allows the organic phase to pass through while the aqueous stream is passed to waste. The purified triflate then combines with a stream of DMF and the material enters a distillation device heated to 70 °C which allows the volatile dichloromethane to be carried out of the reactor with a stream of nitrogen gas. The product then enters a final reactor where it combines with a stream ofalkene and catalyst to form the Heck product. The whole reactor was operated continuously for 5.5 hours, generating approximately 32 mg of product per hour.
 |
||
| Scheme 2 Triflation/Heck coupling facilitated by automated extraction and distillation. |
||
Integration of multiple reaction steps, separations, and purifications into one continuous process has great potential for avoiding energy intensive and wasteful intermediate purification. While great progress has been made, the development of a truly general set of reagents, methods, and devices still requires more research. Immobilized reagents can be wasteful to scale up, and there are significant limitations to current microreactor extraction and distillation technologies. Crystallization is another very important technique in pharmaceutical synthesis, and while there are an increasing number of methods for continuous crystallization,14 15 , it is yet to be used as an intermediate purification step in an automated multi-step synthesis. Lastly, large scale applications of such complex, streamlined processes are required before a thorough assessment of their environmental impact in comparison with traditional batch routes can be made.



- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680
- M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452
- J. G. Kralj, H. R. Sahoo and K. F. Jensen, Lab Chip, 2007, 7, 256–263
- R. L. Hartman, H. R. Sahoo, B. C. Yen and K. F. Jensen, Lab Chip, 2009, 9, 1843–1849
- M. O’Brien, P. Koss, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2012, 10, 7031–7036
- K. K. R. Tetala, J. W. Swarts, B. Chen, A. E. M. Janssen and T. A. van Beek, Lab Chip, 2009, 9, 2085–2092
- D. M. Fries, T. Voitl and P. R. von Rohr, Chem. Eng. Technol., 2008, 31, 1182–1187
- S. Aljbour, H. Yamada and T. Tagawa, Top. Catal., 2010, 53, 694–699
- A. Smirnova, K. Shimura, A. Hibara, M. A. Proskurnin and T. Kitamori, Anal. Sci., 2007, 23, 103–107
- R. C. R. Wootton and A. J. deMello, Chem. Commun., 2004, 266–267
- A. Hibara, K. Toshin, T. Tsukahara, K. Mawatari and T. Kitamora, Chem. Lett., 2008, 1064–1065
- Y. Zhang, S. Kato and T. Anazawa, Lab Chip, 2010, 10, 899–908
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903
- S. Lawton, G. Steele, P. Shering, L. Zhao, I. Laird and X.-W. Ni, Org. Process Res. Dev., 2009, 13, 1357–1363
- H. Zhao, J.-X. Wang, Q.-A. Wang, J.-F. Chen and J. Yun, Ind. Eng. Chem. Res., 2007, 46, 8229–8235
///////

Sorry, the comment form is closed at this time.