











Ragaglitazar
NNC-61-0029, (-) – DRF-2725, NN-622,
(−)DRF 2725
cas 222834-30-2
222834-21-1 (racemate)
Hyperlipidemia; Hypertriglyceridemia; Lipid metabolism disorder; Non-insulin dependent diabetes
PPAR alpha agonist; PPAR gamma agonist
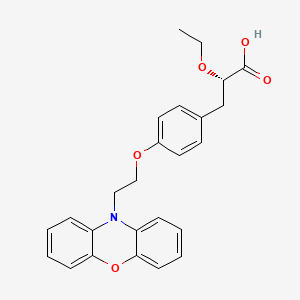
(2S)-2-ETHOXY-3-{4-[2-(10H-PHENOXAZIN-10-YL)ETHOXY]PHENYL}PROPANOIC ACID,
(2S)-2-ethoxy-3-[4-(2-phenoxazin-10-ylethoxy)phenyl]propanoic acid, DRF, 1nyx




……………………………………………………………..

J Med Chem 2001,44(16),2675
http://pubs.acs.org/doi/abs/10.1021/jm010143b

(−)DRF 2725 (6) is a phenoxazine analogue of phenyl propanoic acid. Compound 6 showed interesting dual activation of PPARα and PPARγ. In insulin resistant db/db mice, 6 showed better reduction of plasma glucose and triglyceride levels as compared to rosiglitazone. Compound 6 has also shown good oral bioavailability and impressive pharmacokinetic characteristics. Our study indicates that 6 has great potential as a drug for diabetes and dyslipidemia.

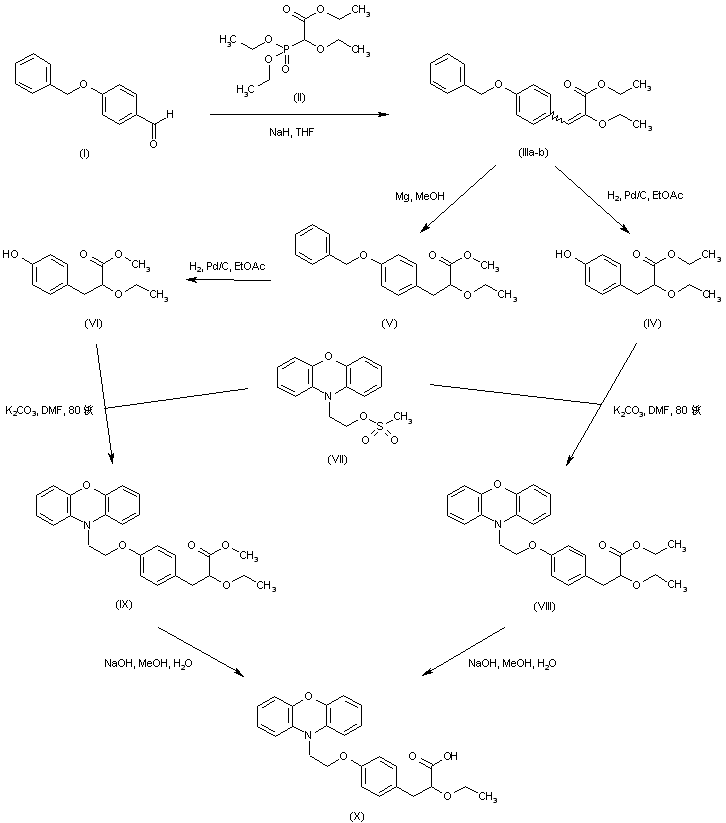
Scheme 1 a
a (a) NaH, DMF, 0−25 °C, 12 h; (b) triethyl 2-ethoxy phosphosphonoacetate, NaH, THF, 0−25 °C, 12 h; (c) Mg/CH3OH, 25 °C, 12 h; (d) 10% aq NaOH, CH3OH, 25 °C, 6 h; (e) (1) pivaloyl chloride, Et3N, DCM, 0 °C, (2) (S)-2-phenyl glycinol/Et3N; (f) 1 M H2SO4, dioxane/water, 90−100 °C, 80 h.
Compound 6 is prepared from phenoxazine using a synthetic route shown in Scheme 1. Phenoxazine 7 upon reaction with p-bromoethoxy benzaldehyde 89 gave benzaldehyde derivative 9. Reacting 9 with triethyl 2-ethoxy phosphonoacetate afforded propenoate 10 as a mixture of geometric isomers. Reduction of 10 using magnesium methanol gave propanoate 11, which on hydrolysis using aqueous sodium hydroxide gave propanoic acid 12 in racemic form. Resolution of 12 using (S)(+)-2-phenyl glycinol followed by hydrolysis using sulfuric acid afforded the propanoic acid 6 in (−) form.
Nate, H.; Matsuki, K.; Tsunashima, A.; Ohtsuka, H.; Sekine, Y. Synthesis of 2-phenylthiazolidine derivatives as cardiotonic agents. II. 2-(phenylpiperazinoalkoxyphenyl)thiazolidine-3-thiocarboxyamides and corresponding carboxamides. Chem. Pharm. Bull. 1987, 35, 2394−2411
(S)-3-[4-[2-(Phenoxazin-10-yl)ethoxy]phenyl]-2-eth-oxypropanoic Acid (6). as a white solid, mp: 89−90 °C.
[α]D 25 = − 12.6 (c = 1.0%, CHCl3).
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J = 7.0 Hz, 3H), 1.42−1.91 (bs, 1H, D2O exchangeable), 2.94−3.15 (m, 2H), 3.40−3.65 (m, 2H), 3.86−4.06 (m, 3H), 4.15 (t, J = 6.6 Hz, 2H), 6.63−6.83 (m, 10H), 7.13 (d, J = 8.5 Hz, 2H). Mass m/z (relative intensity): 419 (M+, 41), 197 (15), 196 (100), 182 (35), 167 (7), 127 (6), 107 (19).
Purity by HPLC: chemical purity: 99.5%; chiral purity: 94.6% (RT 27.5).

…………………………………
http://www.google.com/patents/US6608194?cl=zh
EXAMPLE 23 (−) 3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid:

The title compound (0.19 g, 54%) was prepared as a white solid from diastereomer [(2S-N(1S)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-1-phenyl)ethylpropanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp: 89-90° C.
[α]D 25=−12.6 (c=1.0% CHCl3)
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J=7.02 Hz, 3H), 1.42-1.91 (bs, 1H, D2O exchangeable), 2.94-3.15 (complex, 2H), 3.40-3.65 (complex, 2H), 3.86-4.06 (complex, 3H), 4.15 (t, J=6.65 Hz, 2H), 6.63-6.83 (complex, 10H), 7.13 (d, J=8.54 Hz, 2H).
………………………..
http://www.google.com/patents/EP1049684A1?cl=en
Example 23
(S)-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid :

The title compound (0.19 g, 54 %) was prepared as a white solid from diastereomer [(2S- N(lS)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-l- phenyl)propanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp : 89 – 90 °C. [α]D 25 = – 12.6 (c = 1.0 %, CHC13)
*H NMR (CDC13, 200 MHz) : δ 1.16 (t, J = 7.02 Hz, 3H), 1.42 – 1.91 (bs, IH, D20 exchangeable), 2.94 – 3.15 (complex, 2H), 3.40 – 3.65 (complex, 2H), 3.86 – 4.06 (complex, 3H), 4.15 (t, J = 6.65 Hz, 2H), 6.63 – 6.83 (complex, 10H), 7.13 (d, J = 8.54 Hz, 2H).
| Patent | Submitted | Granted |
|---|---|---|
| Benzamides as ppar modulators [US2006160894] | 2006-07-20 | |
| Novel tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US2002077320] | 2002-06-20 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US7119198] | 2006-07-06 | 2006-10-10 |
| Tricyclic compounds and their use in medicine: process for their preparation and pharmaceutical compositions containing them [US6440961] | 2002-08-27 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6548666] | 2003-04-15 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6608194] | 2003-08-19 | |
| CRYSTALLINE R- GUANIDINES, ARGININE OR (L) -ARGININE (2S) -2- ETHOXY -3-{4- [2-(10H -PHENOXAZIN -10-YL)ETHOXY]PHENYL}PROPANOATE [WO0063189] | 2000-10-26 | |
| Pharmaceutically acceptable salts of phenoxazine and phenothiazine compounds [US6897199] | 2002-11-14 | 2005-05-24 |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6939988] | 2005-09-06 |
WO-2014181362
-
wo/2014/181362 a process for the preparation of 3 … – WIPO
patentscope.wipo.int/search/en/WO2014181362Nov 13, 2014 – (WO2014181362) A PROCESS FOR THE PREPARATION OF 3-ARYL-2-
HYDROXY PROPANOIC ACID COMPOUNDS …
A process for the preparation of 3-aryl-2-hydroxy propanoic acid compounds
ragaglitazar; saroglitazar
Council of Scientific and Industrial Research (India)
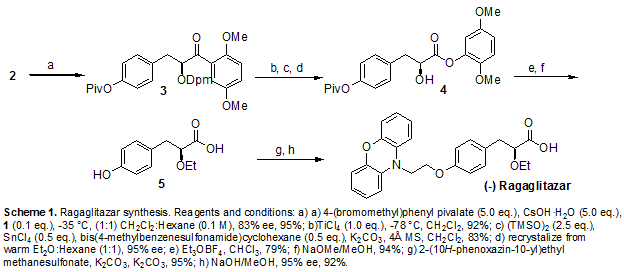
Process for preparing enantiomerically pure 3-aryl-2-hydroxy propanoic acid derivatives (eg ethyl-(S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate), using S-benzyl glycidyl ether as a starting material. Useful as intermediates in the synthesis of peroxisome proliferator activated receptor agonist such as glitazars (eg ragaglitazar or saroglitazar). Appears to be the first filing on these derivatives by the inventors; however see WO2014181359 (for a concurrently published filing) and US8748660 (for a prior filing), claiming synthesis of enantiomerically pure compounds.
- Dolling, U. H.; Davis, P.; Grabowski, E. J. J. Efficient Catalytic Asymmetric Alkylations. 1. Enantioselective Synthesis of (+)-Indacrinone via Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 1984, 106, 446–447.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C. Phase-Transfer Catalyzed Asymmetric Glycolate Alkylation. Org. Lett. 2004, 6, 2289–2292.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C.; Bedke, D. K. Asymmetric Phase-Transfer Catalyzed Glycolate Alkylation, Investigation of the Scope, and Application to the Synthesis of (-)-Ragaglitazar. J. Org. Chem. 2005, ASAP.
- Henke, B. R. Peroxisome Proliferator-Activated Receptor Dual Agonists for the Treatment of Type 2 Diabetes. J. Med. Chem. 2004, 47, 4118–4127.
- Wilson, T. M.; Brown, P. J.; Sternbach, D. D.; Henke, B. R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000, 46, 1306–1317.
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Masuma, R.; Kawakubo, T.; Tanaka, H.; Iwai, Y.; Omura, A. Kurasoins A and B, New Protein Farnesyltrasferase Inhibitors Produced by Paecilomyces sp. FO-3684. J. Antibio. 1996, 49, 932–934.

Sorry, the comment form is closed at this time.