September 4, 2014
FDA Release
The U.S. Food and Drug Administration today granted accelerated approval to Keytruda (pembrolizumab) for treatment of patients with advanced or unresectable melanoma who are no longer responding to other drugs.
Melanoma, which accounts for approximately 5 percent of all new cancers in the United States, occurs when cancer cells form in skin cells that make the pigment responsible for color in the skin. According to the National Cancer Institute, an estimated 76,100 Americans will be diagnosed with melanoma and 9,710 will die from the disease this year.
Keytruda is the first approved drug that blocks a cellular pathway known as PD-1, which restricts the body’s immune system from attacking melanoma cells. Keytruda is intended for use following treatment with ipilimumab, a type of immunotherapy. For melanoma patients whose tumors express a gene mutation called BRAF V600, Keytruda is intended for use after treatment with ipilimumab and a BRAF inhibitor, a therapy that blocks activity of BRAF gene mutations.
“Keytruda is the sixth new melanoma treatment approved since 2011, a result of promising advances in melanoma research,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Many of these treatments have different mechanisms of action and bring new options to patients with melanoma.”
The five prior FDA approvals for melanoma include: ipilimumab (2011), peginterferon alfa-2b (2011), vemurafenib (2011), dabrafenib (2013), and trametinib (2013).
The FDA granted Keytruda breakthrough therapy designation because the sponsor demonstrated through preliminary clinical evidence that the drug may offer a substantial improvement over available therapies. It also received priority review and orphan product designation. Priority review is granted to drugs that have the potential, at the time the application was submitted, to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan product designation is given to drugs intended to treat rare diseases.
The FDA action was taken under the agency’s accelerated approval program, which allows approval of a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients. This program provides earlier patient access to promising new drugs while the company conducts confirmatory clinical trials. An improvement in survival or disease-related symptoms has not yet been established.
Keytruda’s efficacy was established in 173 clinical trial participants with advanced melanoma whose disease progressed after prior treatment. All participants were treated with Keytruda, either at the recommended dose of 2 milligrams per kilogram (mg/kg) or at a higher dose of 10 mg/kg. In the half of the participants who received Keytruda at the recommended dose of 2 mg/kg, approximately 24 percent had their tumors shrink. This effect lasted at least 1.4 to 8.5 months and continued beyond this period in most patients. A similar percentage of patients had their tumor shrink at the 10 mg/kg dose.
Keytruda’s safety was established in the trial population of 411 participants with advanced melanoma. The most common side effects of Keytruda were fatigue, cough, nausea, itchy skin (pruritus), rash, decreased appetite, constipation, joint pain (arthralgia) and diarrhea. Keytruda also has the potential for severe immune-mediated side effects. In the 411 participants with advanced melanoma, severe immune-mediated side effects involving healthy organs, including the lung, colon, hormone-producing glands and liver, occurred uncommonly.
Keytruda is marketed by Merck & Co., based in Whitehouse Station, New Jersey.

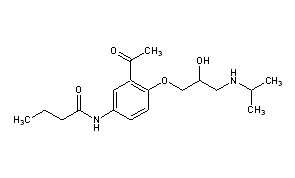
Pembrolizumab, Lambrolizumab, MK-3475
STRUCTURAL FORMULA
Heavy chain
QVQLVQSGVE VKKPGASVKV SCKASGYTFT NYYMYWVRQA PGQGLEWMGG 50
INPSNGGTNF NEKFKNRVTL TTDSSTTTAY MELKSLQFDD TAVYYCARRD 100
YRFDMGFDYW GQGTTVTVSS ASTKGPSVFP LAPCSRSTSE STAALGCLVK 150
DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTKT 200
YTCNVDHKPS NTKVDKRVES KYGPPCPPCP APEFLGGPSV FLFPPKPKDT 250
LMISRTPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY 300
RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT 350
LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS 400
DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE ALHNHYTQKS LSLSLGK 447
Light chain
EIVLTQSPAT LSLSPGERAT LSCRASKGVS TSGYSYLHWY QQKPGQAPRL 50′
LIYLASYLES GVPARFSGSG SGTDFTLTIS SLEPEDFAVY YCQHSRDLPL 100′
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASVVCLL NNFYPREAKV 150′
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200′
THQGLSSPVT KSFNRGEC 218′
Disulfide bridges
22-96 22”-96” 23′-92′ 23”’-92”’ 134-218′ 134”-218”’ 138′-198′ 138”’-198”’
147-203 147”-203” 226-226” 229-229” 261-321 261”-321” 367-425 367”-425”
Glycosylation sites (N)
Asn-297 Asn-297”
lambrolizumab, or MK-3475
| C6504H10004N1716O2036S46 (peptide) | |
| MOL. MASS | 146.3 kDa (peptide) |
Pembrolizumab, Lambrolizumab (also known as MK-3475) is a drug in development by Merck that targets the PD-1 receptor. The drug is intended for use in treating metastatic melanoma.
http://www.ama-assn.org/resources/doc/usan/lambrolizumab.pdf structureof lambrolizumab, or MK-3475
https://download.ama-assn.org/resources/doc/usan/x-pub/pembrolizumab.pdf
Statement on a Nonproprietary Name Adopted by the USAN Council. November 27, 2013.
see above link for change in name
may 2, 2013,
An experimental drug from Merck that unleashes the body’s immune system significantly shrank tumors in 38 percent of patients with advanced melanoma, putting the company squarely in the race to bring to market one of what many experts view as the most promising class of drugs in years.
The drugs are attracting attention here at the annual meeting of the American Society of Clinical Oncology, even though they are still in the early stage of testing. Data from drugs developed by Bristol-Myers Squibb and by Roche had already been released.
The drugs work by disabling a brake that prevents the immune system from attacking cancer cells. The brake is a protein on immune system cells called programmed death 1 receptor, or PD-1.
Merck’s study, which was presented here Sunday and also published in the New England Journal of Medicine, involved 135 patients. While tumors shrank in 38 percent of the patients over all, the rate was 52 percent for patients who got the highest dose of the drug, which is called lambrolizumab, or MK-3475.
But that is what is disclosed tonight, as to pembrolizumab, or MK-3475. Wow. With over $44 billion in 2013 worldwide revenue, that disclosure implies (to seasoned SEC lawyers) that spending on this one drug (or, biologic, to be more technical about it — but remember 40 years ago, Merck had no protein chain biologics research & development programs in its pipe — only chemical drug compounds). . . is material, to that number. Normally that would, in turn, mean that the spending is approaching 5 per cent of revenue. So — Merck may be spending $2.2 billion over the next 12 rolling months, on MK-3475. That’s one BIGhairy science bet, given that Whitehouse Station likely already had over $2 billion invested in the program, at year end 2013.


About Pembrolizumab
Pembrolizumab (MK-3475) is an investigational selective, humanized monoclonal anti-PD-1 antibody designed to block the interaction of PD-1 on T-cells with its ligands, PD-L1 and PD-L2, to reactivate anti-tumor immunity. Pembrolizumab exerts dual ligand blockade of PD-1 pathway.
Today, pembrolizumab is being evaluated across more than 30 types of cancers, as monotherapy and in combination. It is anticipated that by the end of 2014, the pembrolizumab development program will grow to more than 24 clinical trials across 30 different tumor types, enrolling an estimated 6,000 patients at nearly 300 clinical trial sites worldwide, including new Phase 3 studies in head and neck and other cancers. For information about Merck’s oncology clinical studies, please click here.
The Biologics License Application (BLA) for pembrolizumab is under priority review with the U.S. Food and Drug Administration (FDA) for the proposed indication for the treatment of patients with advanced melanoma previously-treated with ipilimumab; the PDUFA date is October 28, 2014. Pembrolizumab has been granted FDA’s Breakthrough Therapy designation for advanced melanoma. If approved by the FDA, pembrolizumab has the potential to be the first PD-1 immune checkpoint modulator approved in this class. The company plans to file a Marketing Authorization Application in Europe for pembrolizumab for advanced melanoma in 2014.
About Head and Neck Cancer
Head and neck cancers are a related group of cancers that involve the oral cavity, pharynx and larynx. Most head and neck cancers are squamous cell carcinomas that begin in the flat, squamous cells that make up the thin surface layer (epithelium) of the head and neck (called the). The leading risk factors for head and neck cancer include tobacco and alcohol use. Infection with certain types of HPV, also called human papillomaviruses, is a risk factor for some types of head and neck cancer, specifically cancer of the oropharynx, which is the middle part of the throat including the soft palate, the base of the tongue, and the tonsils. Each year there are approximately 400,000 cases of cancer of the oral cavity and pharynx, with 160,000 cancers of the larynx, resulting in approximately 300,000 deaths.
About Merck Oncology: A Focus on Immuno-Oncology
At Merck Oncology, our goal is to translate breakthrough science into biomedical innovations to help people with cancer worldwide. Harnessing immune mechanisms to fight cancer is the priority focus of our oncology research and development program. The Company is advancing a pipeline of immunotherapy candidates and combination regimens. Cancer is one of the world’s most urgent unmet medical needs. Helping to empower people to fight cancer is our passion. For information about Merck’s commitment to Oncology visit the Oncology Information Center at http://www.mercknewsroom.com/oncology-infocenter.
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
Hamid, O; Robert, C; Daud, A; Hodi, F. S.; Hwu, W. J.; Kefford, R; Wolchok, J. D.; Hersey, P; Joseph, R. W.; Weber, J. S.; Dronca, R; Gangadhar, T. C.; Patnaik, A; Zarour, H; Joshua, A. M.; Gergich, K; Elassaiss-Schaap, J; Algazi, A; Mateus, C; Boasberg, P; Tumeh, P. C.; Chmielowski, B; Ebbinghaus, S. W.; Li, X. N.; Kang, S. P.; Ribas, A (2013). “Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma”. New England Journal of Medicine 369 (2): 134–44. doi:10.1056/NEJMoa1305133. PMID 23724846

key words
FDA, approved, Keytruda, advanced melanoma, PD-1 blocking drug, pembrolizumab, Lambrolizumab, MK-3475, Monoclonal antibody
















 just an animation
just an animation