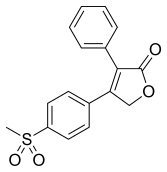
ROFECOXIB
MK-966, MK-0966, Vioxx
162011-90-7
C17-H14-O4-S
314.3596
Rofecoxib /ˌrɒfɨˈkɒksɪb/ is a nonsteroidal anti-inflammatory drug (NSAID) that has now been withdrawn over safety concerns. It was marketed by Merck & Co. to treat osteoarthritis, acute pain conditions, and dysmenorrhoea. Rofecoxib was approved by the Food and Drug Administration (FDA) on May 20, 1999, and was marketed under the brand names Vioxx, Ceoxx, and Ceeoxx.
Rofecoxib gained widespread acceptance among physicians treating patients with arthritis and other conditions causing chronic or acute pain. Worldwide, over 80 million people were prescribed rofecoxib at some time.[1]
On September 30, 2004, Merck withdrew rofecoxib from the market because of concerns about increased risk of heart attack and stroke associated with long-term, high-dosage use. Merck withdrew the drug after disclosures that it withheld information about rofecoxib’s risks from doctors and patients for over five years, resulting in between 88,000 and 140,000 cases of serious heart disease.[2] Rofecoxib was one of the most widely used drugs ever to be withdrawn from the market. In the year before withdrawal, Merck had sales revenue of US$2.5 billion from Vioxx.[3] Merck reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens.
Rofecoxib was available on prescription in both tablet-form and as an oral suspension. It was available by injection for hospital use.
Mode of action
Cyclooxygenase (COX) has two well-studied isoforms, called COX-1 and COX-2. COX-1 mediates the synthesis of prostaglandinsresponsible for protection of the stomach lining, while COX-2 mediates the synthesis of prostaglandins responsible for pain and inflammation. By creating “selective” NSAIDs that inhibit COX-2, but not COX-1, the same pain relief as traditional NSAIDs is offered, but with greatly reduced risk of fatal or debilitating peptic ulcers. Rofecoxib is a selective COX-2 inhibitor, or “coxib”.
Others include Merck’s etoricoxib (Arcoxia), Pfizer’s celecoxib (Celebrex) and valdecoxib (Bextra). Interestingly, at the time of its withdrawal, rofecoxib was the only coxib with clinical evidence of its superior gastrointestinal adverse effect profile over conventional NSAIDs. This was largely based on the VIGOR (Vioxx GI Outcomes Research) study, which compared the efficacy and adverse effect profiles of rofecoxib and naproxen.[4]
Pharmacokinetics
The therapeutic recommended dosages were 12.5, 25, and 50 mg with an approximate bioavailability of 93%.[5][6][7] Rofecoxib crossed the placenta and blood–brain barrier,[5][6][8]and took 1–3 hours to reach peak plasma concentration with an effective half-life (based on steady-state levels) of approximately 17 hours.[5][7][9] The metabolic products are cis-dihydro and trans-dihydro derivatives of rofecoxib[5][9] which are primarily excreted through urine.
Fabricated efficacy studies
On March 11, 2009, Scott S. Reuben, former chief of acute pain at Baystate Medical Center, Springfield, Mass., revealed that data for 21 studies he had authored for the efficacy of the drug (along with others such as celecoxib) had been fabricated in order to augment the analgesic effects of the drugs. There is no evidence that Reuben colluded with Merck in falsifying his data. Reuben was also a former paid spokesperson for the drug company Pfizer (which owns the intellectual property rights for marketing celecoxib in the United States). The retracted studies were not submitted to either the FDA or the European Union’s regulatory agencies prior to the drug’s approval. Drug manufacturer Merckhad no comment on the disclosure.[10]
Adverse drug reactions
Aside from the reduced incidence of gastric ulceration, rofecoxib exhibits a similar adverse effect profile to other NSAIDs.
Prostaglandin is a large family of lipids. Prostaglandin I2/PGI2/prostacyclin is just one member of it. Prostaglandins other than PGI2 (such as PGE2) also play important roles in vascular tone regulation. Prostacyclin/thromboxane are produced by both COX-1 and COX-2, and rofecoxib suppresses just COX-2 enzyme, so there is no reason to believe that prostacyclin levels are significantly reduced by the drug. And there is no reason to believe that only the balance between quantities of prostacyclin and thromboxane is the determinant factor for vascular tone.[11] Indeed Merck has stated that there was no effect on prostacyclin production in blood vessels in animal testing.[12] Other researchers have speculated that the cardiotoxicity may be associated with maleic anhydride metabolites formed when rofecoxib becomes ionized under physiological conditions. (Reddy & Corey, 2005)
Adverse cardiovascular events
VIGOR study and publishing controversy
The VIGOR (Vioxx GI Outcomes Research) study, conducted by Bombardier, et al., which compared the efficacy and adverse effect profiles of rofecoxib and naproxen, had indicated a significant 4-fold increased risk of acute myocardial infarction (heart attack) in rofecoxib patients when compared with naproxen patients (0.4% vs 0.1%, RR 0.25) over the 12 month span of the study. The elevated risk began during the second month on rofecoxib. There was no significant difference in the mortality from cardiovascular events between the two groups, nor was there any significant difference in the rate of myocardial infarction between the rofecoxib and naproxen treatment groups in patients without high cardiovascular risk. The difference in overall risk was by the patients at higher risk of heart attack, i.e. those meeting the criteria for low-dose aspirin prophylaxis of secondary cardiovascular events (previous myocardial infarction, angina, cerebrovascular accident, transient ischemic attack, or coronary artery bypass).
Merck’s scientists interpreted the finding as a protective effect of naproxen, telling the FDA that the difference in heart attacks “is primarily due to” this protective effect (Targum, 2001). Some commentators have noted that naproxen would have to be three times as effective as aspirin to account for all of the difference (Michaels 2005), and some outside scientists warned Merck that this claim was implausible before VIGOR was published.[13] No evidence has since emerged for such a large cardioprotective effect of naproxen, although a number of studies have found protective effects similar in size to those of aspirin.[14][15] Though Dr. Topol’s 2004 paper criticized Merck’s naproxen hypothesis, he himself co-authored a 2001 JAMA article stating “because of the evidence for an antiplatelet effect of naproxen, it is difficult to assess whether the difference in cardiovascular event rates in VIGOR was due to a benefit from naproxen or to a prothrombotic effect from rofecoxib.” (Mukherjee, Nissen and Topol, 2001.)
The results of the VIGOR study were submitted to the United States Food and Drug Administration (FDA) in February 2001. In September 2001, the FDA sent a warning letter to the CEO of Merck, stating, “Your promotional campaign discounts the fact that in the VIGOR study, patients on Vioxx were observed to have a four to five fold increase in myocardial infarctions (MIs) compared to patients on the comparator non-steroidal anti-inflammatory drug (NSAID), Naprosyn (naproxen).”[16] This led to the introduction, in April 2002, of warnings on Vioxx labeling concerning the increased risk of cardiovascular events (heart attack and stroke).
Months after the preliminary version of VIGOR was published in the New England Journal of Medicine, the journal editors learned that certain data reported to the FDA were not included in the NEJM article. Several years later, when they were shown a Merck memo during the depositions for the first federal Vioxx trial, they realized that these data had been available to the authors months before publication. The editors wrote an editorial accusing the authors of deliberately withholding the data.[17] They released the editorial to the media on December 8, 2005, before giving the authors a chance to respond. NEJM editor Gregory Curfman explained that the quick release was due to the imminent presentation of his deposition testimony, which he feared would be misinterpreted in the media. He had earlier denied any relationship between the timing of the editorial and the trial. Although his testimony was not actually used in the December trial, Curfman had testified well before the publication of the editorial.[18]
The editors charged that “more than four months before the article was published, at least two of its authors were aware of critical data on an array of adverse cardiovascular events that were not included in the VIGOR article.” These additional data included three additional heart attacks, and raised the relative risk of Vioxx from 4.25-fold to 5-fold. All the additional heart attacks occurred in the group at low risk of heart attack (the “aspirin not indicated” group) and the editors noted that the omission “resulted in the misleading conclusion that there was a difference in the risk of myocardial infarction between the aspirin indicated and aspirin not indicated groups.” The relative risk for myocardial infarctions among the aspirin not indicated patients increased from 2.25 to 3 (although it remained statitistically insignificant). The editors also noted a statistically significant (2-fold) increase in risk for serious thromboembolic events for this group, an outcome that Merck had not reported in the NEJM, though it had disclosed that information publicly in March 2000, eight months before publication.[19]
The authors of the study, including the non-Merck authors, responded by claiming that the three additional heart attacks had occurred after the prespecified cutoff date for data collection and thus were appropriately not included. (Utilizing the prespecified cutoff date also meant that an additional stroke in the naproxen population was not reported.) Furthermore, they said that the additional data did not qualitatively change any of the conclusions of the study, and the results of the full analyses were disclosed to the FDA and reflected on the Vioxx warning label. They further noted that all of the data in the “omitted” table were printed in the text of the article. The authors stood by the original article.[20]
NEJM stood by its editorial, noting that the cutoff date was never mentioned in the article, nor did the authors report that the cutoff for cardiovascular adverse events was before that for gastrointestinal adverse events. The different cutoffs increased the reported benefits of Vioxx (reduced stomach problems) relative to the risks (increased heart attacks).[19]
Some scientists have accused the NEJM editorial board of making unfounded accusations.[21][22] Others have applauded the editorial. Renowned research cardiologist Eric Topol,[23] a prominent Merck critic, accused Merck of “manipulation of data” and said “I think now the scientific misconduct trial is really fully backed up”.[24] Phil Fontanarosa, executive editor of the prestigious Journal of the American Medical Association, welcomed the editorial, saying “this is another in the long list of recent examples that have generated real concerns about trust and confidence in industry-sponsored studies”.[25]
On May 15, 2006, the Wall Street Journal reported that a late night email, written by an outside public relations specialist and sent to Journal staffers hours before the Expression of Concern was released, predicted that “the rebuke would divert attention to Merck and induce the media to ignore the New England Journal of Medicine‘s own role in aiding Vioxx sales.”[26]
“Internal emails show the New England Journal’s expression of concern was timed to divert attention from a deposition in which Executive Editor Gregory Curfman made potentially damaging admissions about the journal’s handling of the Vioxx study. In the deposition, part of the Vioxx litigation, Dr. Curfman acknowledged that lax editing might have helped the authors make misleading claims in the article.” The Journal stated that NEJM‘s “ambiguous” language misled reporters into incorrectly believing that Merck had deleted data regarding the three additional heart attacks, rather than a blank table that contained no statistical information; “the New England Journal says it didn’t attempt to have these mistakes corrected.”[26]
Alzheimer’s studies
In 2000 and 2001, Merck conducted several studies of rofecoxib aimed at determining if the drug slowed the onset of Alzheimer’s disease. Merck has placed great emphasis on these studies on the grounds that they are relatively large (almost 3000 patients) and compared rofecoxib to a placebo rather than to another pain reliever. These studies found an elevated death rate among rofecoxib patients, although the deaths were not generally heart-related. However, they did not find any elevated cardiovascular risk due to rofecoxib.[27] Before 2004, Merck cited these studies as providing evidence, contrary to VIGOR, of rofecoxib’s safety.
APPROVe study
In 2001, Merck commenced the APPROVe (Adenomatous Polyp PRevention On Vioxx) study, a three-year trial with the primary aim of evaluating the efficacy of rofecoxib for theprophylaxis of colorectal polyps. Celecoxib had already been approved for this indication, and it was hoped to add this to the indications for rofecoxib as well. An additional aim of the study was to further evaluate the cardiovascular safety of rofecoxib.
The APPROVe study was terminated early when the preliminary data from the study showed an increased relative risk of adverse thrombotic cardiovascular events (includingheart attack and stroke), beginning after 18 months of rofecoxib therapy. In patients taking rofecoxib, versus placebo, the relative risk of these events was 1.92 (rofecoxib 1.50 events vs placebo 0.78 events per 100 patient years). The results from the first 18 months of the APPROVe study did not show an increased relative risk of adverse cardiovascular events. Moreover, overall and cardiovascular mortality rates were similar between the rofecoxib and placebo populations.[28]
In summary, the APPROVe study suggested that long-term use of rofecoxib resulted in nearly twice the risk of suffering a heart attack or stroke compared to patients receiving a placebo.
Other studies
Pre-approval Phase III clinical trials, like the APPROVe study, showed no increased relative risk of adverse cardiovascular events for the first eighteen months of rofecoxib usage (Merck, 2004). Others have pointed out that “study 090,” a pre-approval trial, showed a 3-fold increase in cardiovascular events compared to placebo, a 7-fold increase compared to nabumetone (another [NSAID]), and an 8-fold increase in heart attacks and strokes combined compared to both control groups.[29][30] Although this was a relatively small study and only the last result was statistically significant, critics have charged that this early finding should have prompted Merck to quickly conduct larger studies of rofecoxib’s cardiovascular safety. Merck notes that it had already begun VIGOR at the time Study 090 was completed. Although VIGOR was primarily designed to demonstrate new uses for rofecoxib, it also collected data on adverse cardiovascular outcomes.
Several very large observational studies have also found elevated risk of heart attack from rofecoxib. For example, a recent retrospective study of 113,000 elderly Canadians suggested a borderline statistically significant increased relative risk of heart attacks of 1.24 from Vioxx usage, with a relative risk of 1.73 for higher-dose Vioxx usage. (Levesque, 2005). Another study, using Kaiser Permanente data, found a 1.47 relative risk for low-dose Vioxx usage and 3.58 for high-dose Vioxx usage compared to current use of celecoxib, though the smaller number was not statistically significant, and relative risk compared to other populations was not statistically significant. (Graham, 2005).
Furthermore, a more recent meta-study of 114 randomized trials with a total of 116,000+ participants, published in JAMA, showed that Vioxx uniquely increased risk of renal (kidney) disease, and heart arrhythmia.[31]
Other COX-2 inhibitors
Any increased risk of renal and arrhythmia pathologies associated with the class of COX-2 inhibitors, e.g. celecoxib (Celebrex), valdecoxib (Bextra), parecoxib (Dynastat),lumiracoxib, and etoricoxib is not evident,[31] although smaller studies[32][33] had demonstrated such effects earlier with the use of celecoxib, valdecoxib and parecoxib.
Nevertheless, it is likely that trials of newer drugs in the category will be extended in order to supply additional evidence of cardiovascular safety. Examples are some more specific COX-2 inhibitors, including etoricoxib (Arcoxia) and lumiracoxib (Prexige), which are currently (circa 2005) undergoing Phase III/IV clinical trials.
Besides, regulatory authorities worldwide now require warnings about cardiovascular risk of COX-2 inhibitors still on the market. For example, in 2005, EU regulators required the following changes to the product information and/or packaging of all COX-2 inhibitors:[34]
- Contraindications stating that COX-2 inhibitors must not be used in patients with established ischaemic heart disease and/or cerebrovascular disease (stroke), and also in patients with peripheral arterial disease
- Reinforced warnings to healthcare professionals to exercise caution when prescribing COX-2 inhibitors to patients with risk factors for heart disease, such as hypertension, hyperlipidaemia (high cholesterol levels), diabetes and smoking
- Given the association between cardiovascular risk and exposure to COX-2 inhibitors, doctors are advised to use the lowest effective dose for the shortest possible duration of treatment
Other NSAIDs
Since the withdrawal of Vioxx it has come to light that there may be negative cardiovascular effects with not only other COX-2 inhibitiors, but even the majority of other NSAIDs. It is only with the recent development of drugs like Vioxx that drug companies have carried out the kind of well executed trials that could establish such effects and these sort of trials have never been carried out in older “trusted” NSAIDs such as ibuprofen, diclofenac and others. The possible exceptions may be aspirin and naproxen due to their anti-platelet aggregation properties.
Withdrawal
Due to the findings of its own APPROVe study, Merck publicly announced its voluntary withdrawal of the drug from the market worldwide on September 30, 2004.[35]
In addition to its own studies, on September 23, 2004 Merck apparently received information about new research by the FDA that supported previous findings of increased risk of heart attack among rofecoxib users (Grassley, 2004). FDA analysts estimated that Vioxx caused between 88,000 and 139,000 heart attacks, 30 to 40 percent of which were probably fatal, in the five years the drug was on the market.[36]
On November 5, the medical journal The Lancet published a meta-analysis of the available studies on the safety of rofecoxib (Jüni et al., 2004). The authors concluded that, owing to the known cardiovascular risk, rofecoxib should have been withdrawn several years earlier. The Lancet published an editorial which condemned both Merck and the FDA for the continued availability of rofecoxib from 2000 until the recall. Merck responded by issuing a rebuttal of the Jüni et al. meta-analysis that noted that Jüni omitted several studies that showed no increased cardiovascular risk. (Merck & Co., 2004).
In 2005, advisory panels in both the U.S. and Canada encouraged the return of rofecoxib to the market, stating that rofecoxib’s benefits outweighed the risks for some patients. The FDA advisory panel voted 17-15 to allow the drug to return to the market despite being found to increase heart risk. The vote in Canada was 12-1, and the Canadian panel noted that the cardiovascular risks from rofecoxib seemed to be no worse than those from ibuprofen—though the panel recommended that further study was needed for all NSAIDs to fully understand their risk profiles. Notwithstanding these recommendations, Merck has not returned rofecoxib to the market.[37]
In 2005, Merck retained Debevoise & Plimpton LLP to investigate Vioxx study results and communications conducted by Merck. Through the report, it was found that Merck’s senior management acted in good faith, and that the confusion over the clinical safety of Vioxx was due to the sales team’s overzealous behavior. The report that was filed gave a timeline of the events surrounding Vioxx and showed that Merck intended to operate honestly throughout the process. Any mistakes that were made regarding the mishandling of clinical trial results and withholding of information was the result of oversight, not malicious behavior. The Martin Report did conclude that the Merck’s marketing team exaggerated the safety of Vioxx and replaced truthful information with sales tactics.[citation needed] The report was published in February 2006, and Merck was satisfied with the findings of the report and promised to consider the recommendations contained in the Martin Report. Advisers to the US Food and Drug Administration (FDA) have voted, by a narrow margin, that it should not ban Vioxx — the painkiller withdrawn by drug-maker Merck.
They also said that Pfizer’s Celebrex and Bextra, two other members of the family of painkillers known as COX-2 inhibitors, should remain available, despite the fact that they too boost patients’ risk of heart attack and stroke. url = http://www.nature.com/drugdisc/news/articles/433790b.html The recommendations of the arthritis and drug safety advisory panel offer some measure of relief to the pharmaceutical industry, which has faced a barrage of criticism for its promotion of the painkillers. But the advice of the panel, which met near Washington DC over 16–18 February, comes with several strings attached.
For example, most panel members said that manufacturers should be required to add a prominent warning about the drugs’ risks to their labels; to stop direct-to-consumer advertising of the drugs; and to include detailed, written risk information with each prescription. The panel also unanimously stated that all three painkillers “significantly increase the risk of cardiovascular events”.
The panel voted 17 to 15 against banning Vioxx (rofecoxib) entirely; the vote on Bextra (valdecoxib) was 17 to 13 with 2 abstentions; Celebrex (celecoxib) was endorsed 31 to 1. Shares of Merck, based in Whitehouse Station, New Jersey, and New York-based Pfizer closed up 13% and 7% respectively on 18 February, 2013, the day of the votes.
The FDA is expected to act on the recommendations within weeks. Although the agency usually follows the recommendations of its outside advisers, it is not bound to do so. A top official said that, in light of the closeness of some of the votes, the agency will examine the panel members’ comments in detail before deciding what to do.
An official from Merck said during the meeting that it would consider reintroducing Vioxx, which it withdrew in September 2004. On April 7, 2005, Pfizer withdrew Bextra from the U.S. market on recommendation by the FDA. Pfizer’s other painkiller, Celebrex, is still on the market.
Litigation
As of March 2006, there had been over 10,000 cases and 190 class actions filed against Merck[citation needed] over adverse cardiovascular events associated with rofecoxib and the adequacy of Merck’s warnings. The first wrongful death trial, Rogers v. Merck, was scheduled in Alabama in the spring of 2005, but was postponed after Merck argued that the plaintiff had falsified evidence of rofecoxib use.[1]
On August 19, 2005, a jury in Texas voted 10-2 to hold Merck liable for the death of Robert Ernst, a 59-year-old man who allegedly died of a rofecoxib-induced heart attack. The plaintiffs’ lead attorney was Mark Lanier. Merck argued that the death was due to cardiac arrhythmia, which had not been shown to be associated with rofecoxib use. The jury awarded Carol Ernst, widow of Robert Ernst, $253.4 million in damages. This award will almost certainly be capped at no more than US$26.1 million because of punitive damages limits under Texas law.[2] As of March 2006, the plaintiff had yet to ask the court to enter a judgment on the verdict; Merck has stated that it will appeal.
On November 3, 2005, Merck won the second case Humeston v. Merck, a personal injury case, in Atlantic City, New Jersey. The plaintiff experienced a mild myocardial infarction and claimed that rofecoxib was responsible, after having taken it for two months. Merck argued that there was no evidence that rofecoxib was the cause of Humeston’s injury and that there is no scientific evidence linking rofecoxib to cardiac events with short durations of use. The jury ruled that Merck had adequately warned doctors and patients of the drug’s risk.[3]
The first federal trial on rofecoxib, Plunkett v. Merck, began on November 29, 2005 in Houston. The trial ended in a hung jury and a mistrial was declared on December 12, 2005. According to the Wall Street Journal, the jury hung by an eight to one majority, favoring the defense. Upon retrial in February 2006 in New Orleans, where the Vioxx multidistrict litigation (MDL) is based, a jury found Merck not liable, even though the plaintiffs had the NEJM editor testify as to his objections to the VIGOR study.
On January 30, 2006, a New Jersey state court dismissed a case brought by Edgar Lee Boyd, who blamed Vioxx for gastrointestinal bleeding that he experienced after taking the drug. The judge said that Boyd failed to prove the drug caused his stomach pain and internal bleeding.
In January 2006, Garza v. Merck began trial in Rio Grande City, Texas. The plaintiff, a 71-year-old smoker with heart disease, had a fatal heart attack three weeks after finishing a one-week sample of rofecoxib. On April 21, 2006 the jury awarded the plaintiff $7 million compensatory and $25 million punitive. The Texas state court of appeals in San Antonio later rules Garza’s fatal heart attack probably resulted from pre-existing health conditions unrelated to his taking of Vioxx, thus reversing the $32 million jury award.[4]
On April 5, 2006, the jury held Merck liable for the heart attack of 77-year-old John McDarby, and awarded Mr McDarby $4.5 million in compensatory damages based on Merck’s failure to properly warn of Vioxx safety risks. After a hearing on April 11, 2006, the jury also awarded Mr McDarby an additional $9 million in punitive damages. The same jury found Merck not liable for the heart attack of 60-year-old Thomas Cona, a second plaintiff in the trial, but was liable for fraud in the sale of the drug to Cona.
Merck has reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens. Patients who claim to have suffered as a result of taking Vioxx in countries outside the US are campaigning for this to be extended.
In March 2010, an Australian class-action lawsuit against Merck ruled that Vioxx doubled the risk of heart attacks, and that Merck had breached the Trade Practices Act by selling a drug which was unfit for sale.[38]
In November 2011, Merck announced a civil settlement with the US Attorney’s Office for the District of Massachusetts, and individually with 43 US states and the District of Columbia, to resolve civil claims relating to Vioxx.[5] Under the terms of the settlement, Merck agreed to pay two-thirds of a previously recorded $950 million reserve charge in exchange for release from civil liability. Litigation with seven additional states remains outstanding. Under separate criminal proceedings, Merck plead guilty to a federal misdemeanor charge relating to the marketing of the drug across state lines, incurring a fine of $321.6 million.[6]
Other effects
Rofecoxib was shown to improve premenstrual acne vulgaris in a placebo controlled study.[39]
Synthesis

Rofecoxib synthesis.[40]
,,,,,,,,,,,,,,,,,
The oxidation of 4- (methylsulfanyl) acetophenone (X) with monoperoxyphthalic acid (MMPP) in dichloro-methane / methanol gives the corresponding sulfone (XI), which is brominated with Br2 / AlCl3 in chloroform, yielding the expected phenacyl bromide ( XII). Finally, this compound is cyclocondensed with phenylacetic acid (I) by means of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) and triethylamine in acetonitrile. 5) Reaction of [4- (methylsulfonyl ) phenyl] phenylacetyl-ene (XIII) with CO catalyzed by Rh4 (CO) 12 in THF at 100 C in a stainless steel autoclave at 100 Atm pressure, followed by a chromatographic separation in a silicagel column to eliminate the undesired regioisomer.

……………….
The synthesis of rofecoxib can be performed by several different ways: 1) The condensation of phenylacetic acid (I) with ethyl bromoacetate (II) by means of triethylamine in THF yields 2- (phenylacetoxy) acetic acid ethyl ester (III), which is cyclized to the hydroxyfuranone (IV) by means of potassium tert-butoxide in tert-butanol. The reaction of (IV) with triflic anhydride and diisopropylethylamine in dichloro-methane affords the corresponding triflate (V), which by reaction with LiBr in hot acetone yields the bromofuranone (VI) The condensation of (VI) with 4- (methylsulfanyl) phenylboronic acid (VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene gives 4- [4- (methylsulfanyl) -phenyl]. – 3-phenylfuran-2 (5H) -one (VIII), which is finally oxidized with 2KHSO5.KHSO4.K2SO4 (oxone). 2) The intermediate (VIII) can also be obtained by condensation of triflate (V) with boronic acid ( VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene. 3) The intermediate (VIII) can also be synthesized by the reaction of triflate (V) with tetramethylammonium chloride, giving the chlorofuranone (IX), which is then condensed with boronic acid (VII) as before.

Footnotes
- Jump up^ http://www.npr.org/templates/story/story.php?storyId=4054991
- Jump up^ “Up to 140,000 heart attacks linked to Vioxx.”. New Scientist. 2005-01-25. p. 1.
- Jump up^ “Merck Sees Slightly Higher 2007 Earnings”. New York Times. Reuters. 2006-12-07. p. A1.
- Jump up^ Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M. B.; Hawkey, C. J.; Hochberg, M. C.; Kvien, T. K.; Schnitzer, T. J.; Vigor Study, G. (2000). “Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis”. New England Journal of Medicine 343 (21): 1520–1528, 2 1528 following 1528. doi:10.1056/NEJM200011233432103. PMID 11087881. edit
- ^ Jump up to:a b c d Merck & Co. VIOXX (rofecoxib tablets and oral suspension). Accessed at: http://www.merck.com/product/usa/pi_circulars/v/vioxx/vioxx_pi.pdf 01 Feb 2010
- ^ Jump up to:a b Gold Standard Inc. Rofecoxib Vioxx Accessed at: http://www.mdconsult.com/das/pharm/body/181267313-3/946823742/full/2399 01 Feb 2010
- ^ Jump up to:a b Davies, N. M.; Teng, X. W.; Skjodt, N. M. (2003). “Pharmacokinetics of rofecoxib: a specific cyclo-oxygenase-2 inhibitor”. Clinical pharmacokinetics 42 (6): 545–556.PMID 12793839. edit
- Jump up^ Padi, S.; Kulkarni, S. (2004). “Differential effects of naproxen and rofecoxib on the development of hypersensitivity following nerve injury in rats”. Pharmacology, Biochemistry, and Behavior 79 (2): 349–358. doi:10.1016/j.pbb.2004.08.005. PMID 15501312. edit
- ^ Jump up to:a b Scott, L. J.; Lamb, H. M. (1999). “Rofecoxib”. Drugs 58 (3): 499–505; discussion 506–7. doi:10.2165/00003495-199958030-00016. PMID 10493277. edit
- Jump up^ Winstein, Keith J. (March 11, 2009). “Top Pain Scientist Fabricated Data in Studies, Hospital Says”. The Wall Street Journal.
- Jump up^ Vane, J.; Bakhle, Y.; Botting, R. (1998). “Cyclooxygenases 1 and 2”. Annual review of pharmacology and toxicology 38: 97–120. doi:10.1146/annurev.pharmtox.38.1.97.PMID 9597150. edit
- Jump up^ sfgate.com
- Jump up^ www.saferdrugsnow.org
- Jump up^ Karha, J.; Topol, E. J. (2004). “The sad story of Vioxx, and what we should learn from it”. Cleveland Clinic journal of medicine 71 (12): 933–934, 936, 934–9.doi:10.3949/ccjm.71.12.933. PMID 15641522. edit
- Jump up^ Solomon, D. H.; Glynn, R. J.; Levin, R.; Avorn, J. (2002). “Nonsteroidal anti-inflammatory drug use and acute myocardial infarction”. Archives of Internal Medicine 162 (10): 1099–1104.doi:10.1001/archinte.162.10.1099. PMID 12020178. edit
- Jump up^http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM166383.pdf
- Jump up^ Curfman, G.; Morrissey, S.; Drazen, J. (2005). “Expression of concern: Bombardier et al., “Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis,” N Engl J Med 2000;343:1520-8″. The New England Journal of Medicine 353 (26): 2813–2814. doi:10.1056/NEJMe058314. PMID 16339408. edit
- Jump up^ http://www.forbes.com/work/feeds/ap/2006/02/13/ap2523250.html. Missing or empty
|title= (help)[dead link]
- ^ Jump up to:a b Curfman, G.; Morrissey, S.; Drazen, J. (2006). “Expression of concern reaffirmed”. The New England Journal of Medicine 354 (11): 1193. doi:10.1056/NEJMe068054.PMID 16495386. edit
- Jump up^ Bombardier, C.; Laine, L.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.; Hawkey, C.; Hochberg, M.; Kvien, T.; Schnitzer, T. J.; Weaver, A. (2006). “Response to expression of concern regarding VIGOR study”. The New England Journal of Medicine 354 (11): 1196–1199. doi:10.1056/NEJMc066096. PMID 16495387. edit
- Jump up^ http://pipeline.corante.com/archives/2006/02/22/nejm_vs_its_contributors_round_two.php
- Jump up^ http://dimer.tamu.edu/simplog/archive.php?blogid=3&pid=3293
- Jump up^ http://genetics.case.edu/faculty2.php?fac=ejt9
- Jump up^ http://www.medicinenet.com/script/main/art.asp?articlekey=56384&page=2
- Jump up^ http://www.beasleyallen.com/news/vioxx-plaintiffs-seek-mistrial-after-allegation-on-merck-study/
- ^ Jump up to:a b David Armstrong (2006-05-15). “How the New England Journal Missed Warning Signs on Vioxx”. Wall Street Journal. p. A1.
- Jump up^ Konstam, M. A.; Weir, M. R.; Reicin, A.; Shapiro, D.; Sperling, R. S.; Barr, E.; Gertz, B. J. (2001). “Cardiovascular thrombotic events in controlled, clinical trials of rofecoxib”. Circulation104 (19): 2280–2288. doi:10.1161/hc4401.100078. PMID 11696466. edit
- Jump up^ Bresalier, R.; Sandler, R.; Quan, H.; Bolognese, J.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; Konstam, M. A.; Baron, J. A.; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators (2005). “Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial”. The New England Journal of Medicine 352(11): 1092–1102. doi:10.1056/NEJMoa050493. PMID 15713943. edit
- Jump up^ http://www.fda.gov/ohrms/dockets/ac/01/briefing/3677b2_06_cardio.pdf
- Jump up^ Wolfe, M. M. (2004). “Rofecoxib, Merck, and the FDA”. The New England Journal of Medicine 351 (27): 2875–2878; author 2878 2875–2878. doi:10.1056/NEJM200412303512719.PMID 15625749. edit
- ^ Jump up to:a b Zhang, J.; Ding, E.; Song, Y. (2006). “Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials”. Journal of the American Medical Association 296 (13): 1619–1632. doi:10.1001/jama.296.13.jrv60015. PMID 16968832. edit
- Jump up^ Solomon, S.; McMurray, J.; Pfeffer, M.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.; Zauber, A.; Hawk, E.; Bertagnolli, M.; Adenoma Prevention with Celecoxib (APC) Study Investigators (2005). “Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention”. The New England Journal of Medicine 352 (11): 1071–1080.doi:10.1056/NEJMoa050405. PMID 15713944. edit
- Jump up^ Nussmeier, N.; Whelton, A.; Brown, M.; Langford, R.; Hoeft, A.; Parlow, J.; Boyce, S.; Verburg, K. (2005). “Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery”. The New England Journal of Medicine 352 (11): 1081–1091. doi:10.1056/NEJMoa050330. PMID 15713945. edit
- Jump up^ “European Medicines Agency concludes action on COX-2 inhibitors” (pdf). European Medicines Agency. Retrieved 2008-04-16.
- Jump up^ “Merck Announces Voluntary Worldwide Withdrawal of VIOXX” (pdf). Retrieved 2008-04-16.
- Jump up^ “Congress Questions Vioxx, FDA”. PBS NewsHour. 2004-11-18. Retrieved 2013-06-03.
- Jump up^ “SUMMARY: Report of the Expert Advisory Panel on the Safety of Cox-2 Selective Non-steroidal Anti-Inflammatory Drugs (NSAIDs)”. Health Canada. 2005-07-06. Retrieved 2011-06-04.
- Jump up^ Drug unfit for sale, says judge in compo case The Age, March 6, 2010
- Jump up^ http://bioline.utsc.utoronto.ca/archive/00002693/01/dv04120.pdf#search=%22acne%20rofecoxib%22
- Jump up^ http://vioxxlawyer.org/rofecoxib-synthesis/
References
- FDA (2005). “Summary minutes for the February 16, 17 and 18, 2005, Joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee.” Published on the internet, March 2005. Link
- Fitzgerald GA, Coxibs and Cardiovascular Disease, N Engl J Med 2004;351(17): 1709–1711. PMID 15470192.
- Grassley CE (15 Oct 2004). Grassley questions Merck about communication with the FDA on Vioxx. Press Release.
- Jüni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M (2004). Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet (published online; see also Merck response below)
- Karha J and Topol EJ. The sad story of Vioxx, and what we should learn from it Cleve Clin J Med 2004; 71(12):933-939. PMID 15641522
- Michaels, D. (June 2005) DOUBT Is Their Product, Scientific American, 292 (6).
- Merck & Co., (5 Nov 2004). Response to Article by Juni et al. Published in The Lancet on Nov. 5. Press Release.
- Merck & Co (30 Sep 2004) Merck Announces Voluntary Worldwide Withdrawal of VIOXX. Press release [7].
- D. M. Mukherjee, S. E. Nissen, and E. J. Topol, “Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors,” Journal of the American Medical Association 186 (2001): 954–959.
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352(11):1081-91. PMID 15713945
- Okie, S (2005) “Raising the safety bar–the FDA’s coxib meeting.” N Engl J Med. 2005 Mar 31;352(13):1283-5. PMID 15800221.
- Leleti Rajender Reddy, Corey EJ. Facile air oxidation of the conjugate base of rofecoxib (Vioxx), a possible contributor to chronic human toxicity Tetrahedron Lett 2005, 46: 927. doi:10.1016/j.tetlet.2004.12.055
- Swan SK et al., Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. Annals of Int Med 2000; 133:1–9
- Targum, SL. (1 Feb. 2001) Review of cardiovascular safety database. FDA memorandum. [8]
- Wolfe, MM et al., Gastrointestinal Toxicity of Nonsteroidal Anti-anflamattory Drugs, New England Journal of Medicine. 1999; 340; 1888-98.
External links

















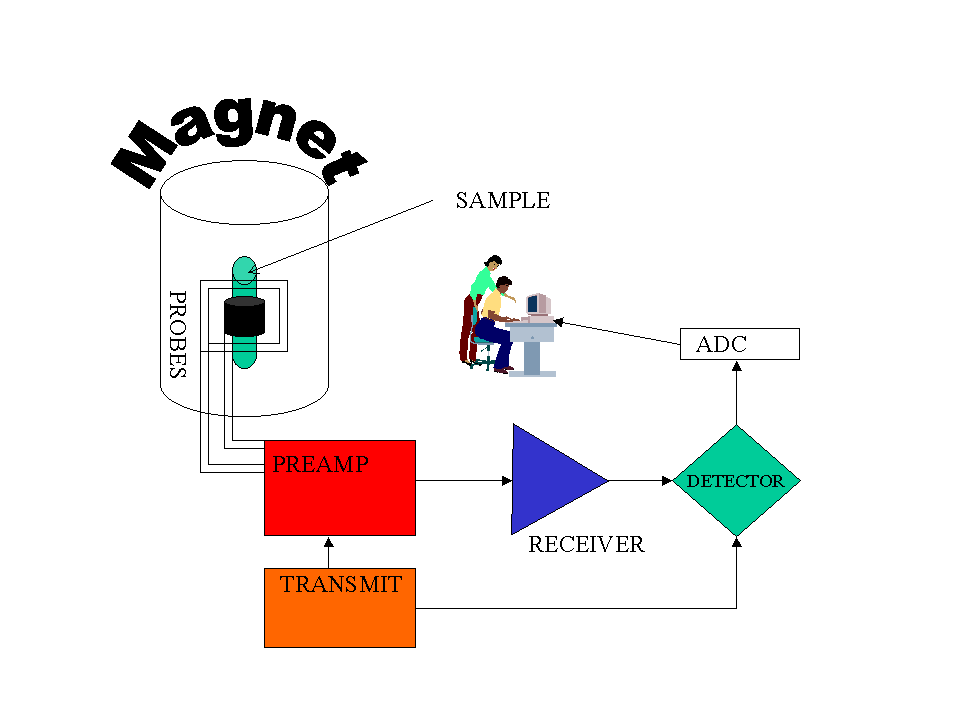












 Dedicated to all moms
Dedicated to all moms

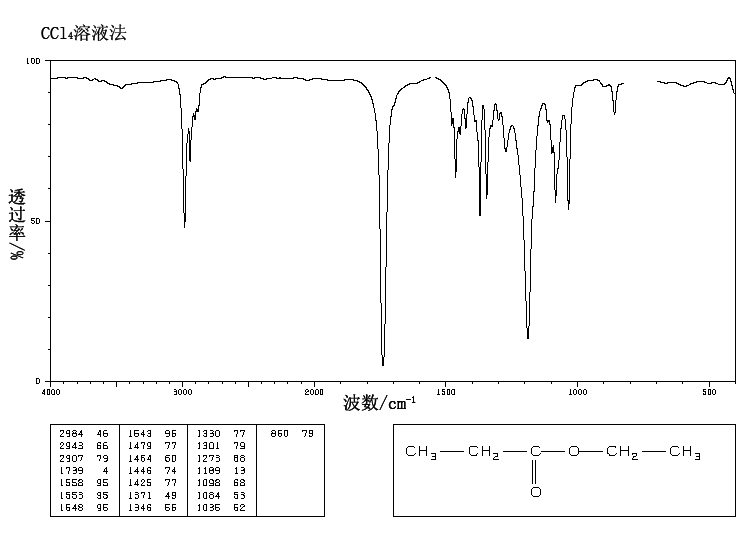
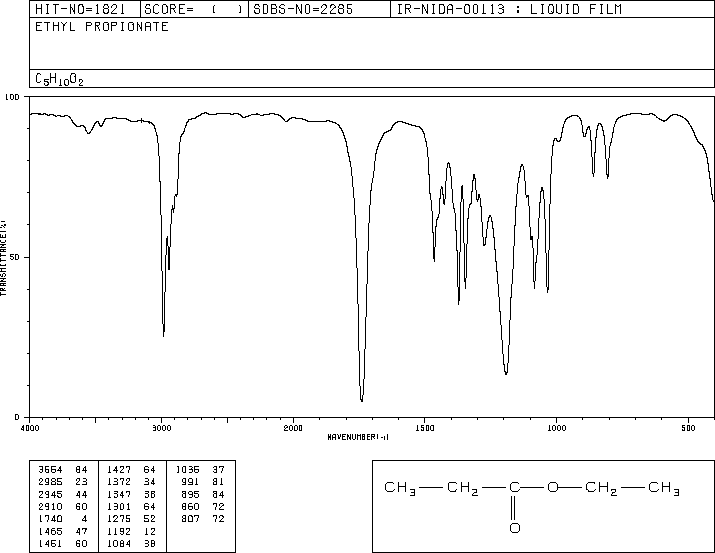


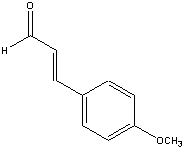
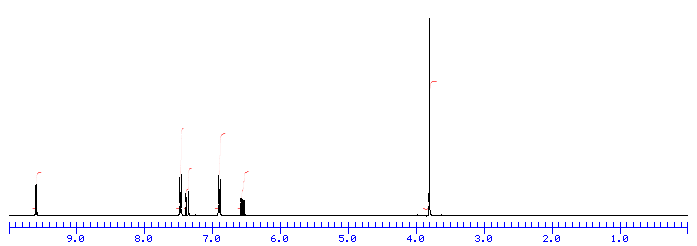
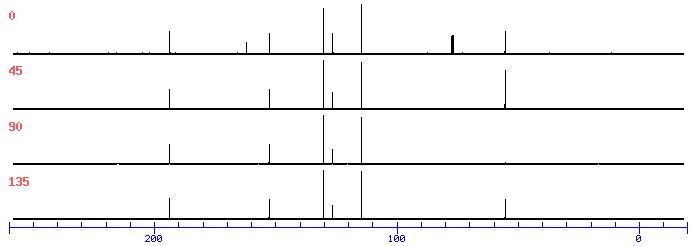
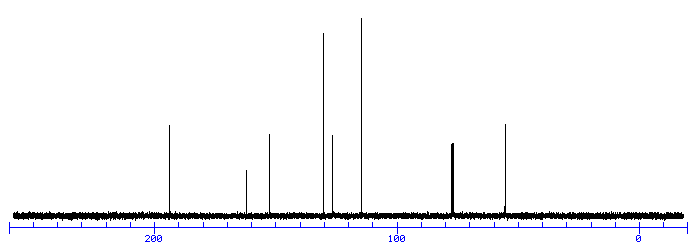
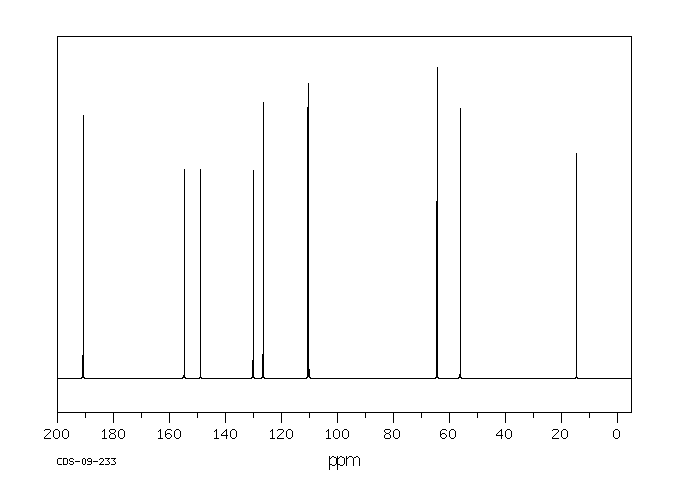
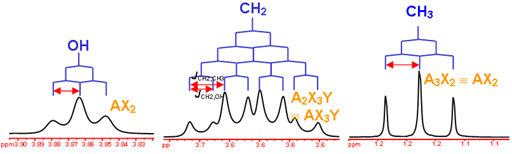
 This is the structure. See if you can assign the peaks on your own.
This is the structure. See if you can assign the peaks on your own. C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.
C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.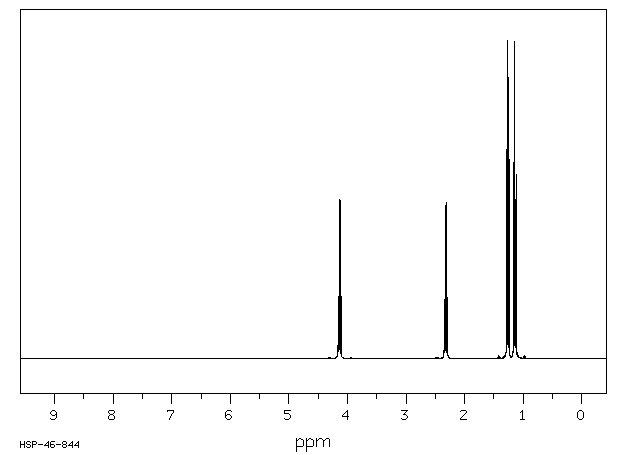

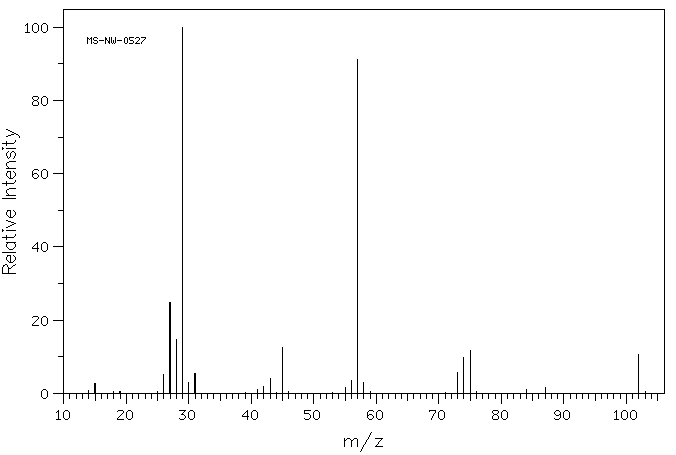
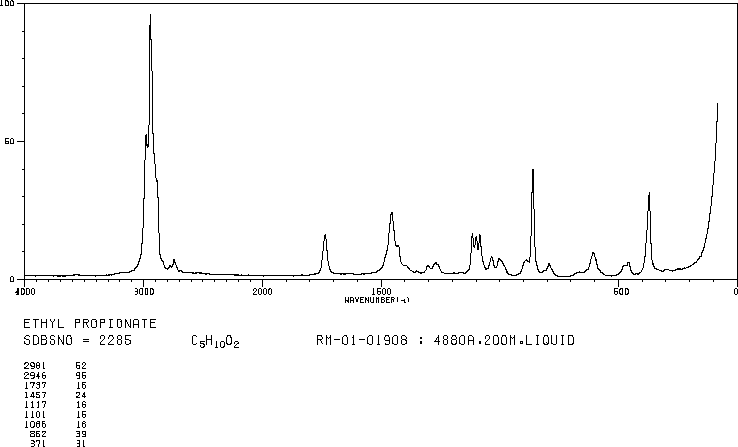


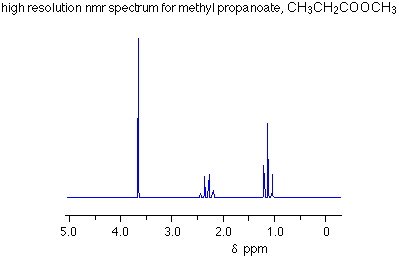

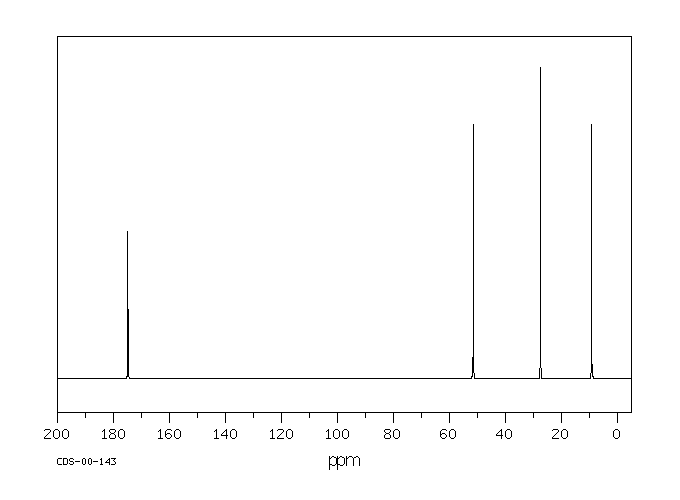



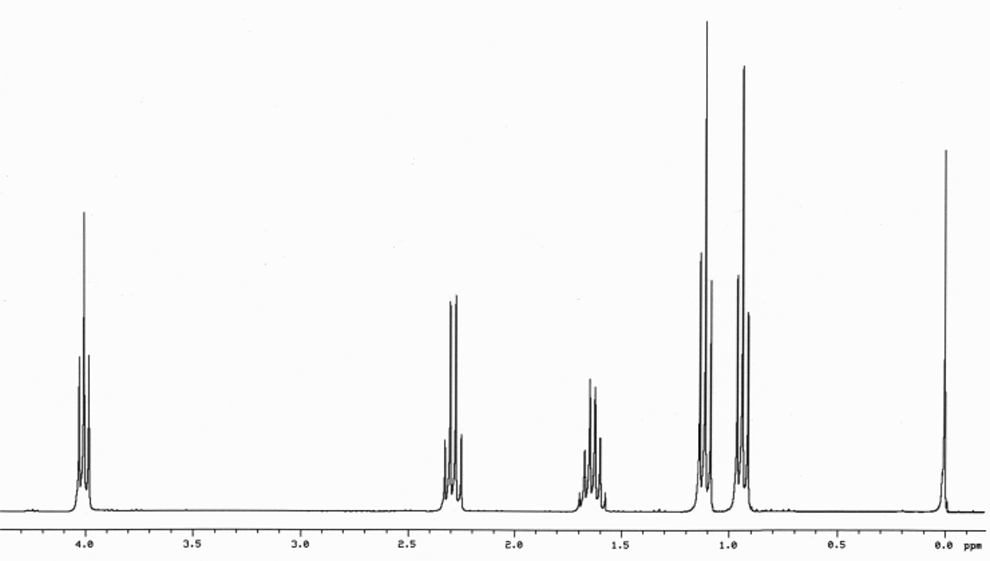












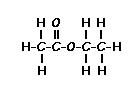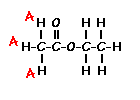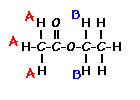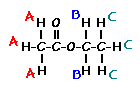
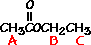


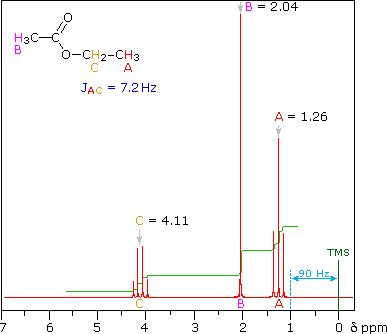


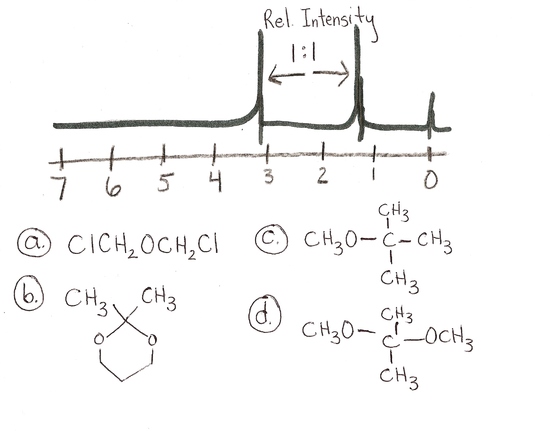 answer is a and d
answer is a and d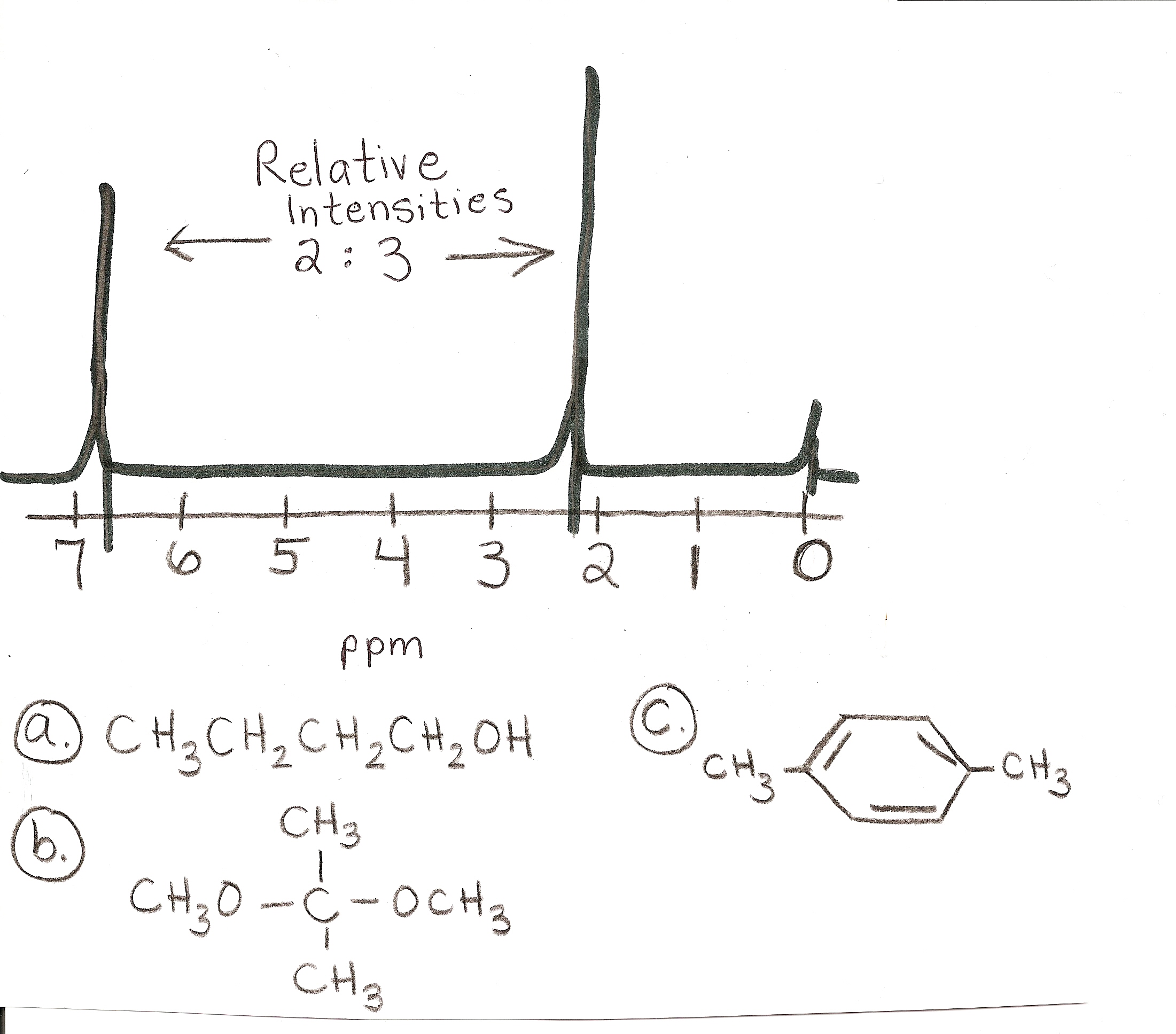
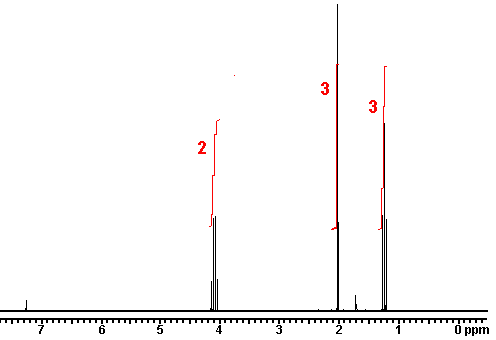
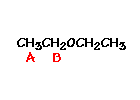
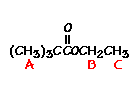




 n
n



