











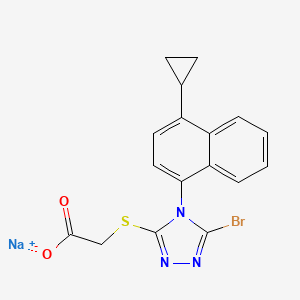
Lesinurad
Acetic acid, 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]-,
sodium salt (1:1)
Sodium 2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-
yl]sulfanyl}acetate
2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid
MOLECULAR FORMULA C17H13BrN3NaO2S
MOLECULAR WEIGHT 426.3
http://clinicaltrials.gov/show/NCT01508702
http://www.ama-assn.org/resources/doc/usan/lesinurad.pdf
Ardea Biosciences, Inc.
- Lesinurad
- RDEA 594
- RDEA594
- UNII-09ERP08I3W
Gout phase 3
Gout is associated with elevated levels of uric acid that crystallize and deposit in joints, tendons, and surrounding tissues. Gout is marked by recurrent attacks of red, tender, hot, and/or swollen joints.
This study will assess the serum uric acid lowering effects and safety of lesinurad compared to placebo in patients who are intolerant or have a contraindication to allopurinol or febuxostat.
http://euroscan.org.uk/technologies/technology/view/2386
Lesinurad (RDEA-594, lesinurad sodium) is a selective urate transporter-1 (URAT-1) inhibitor, which blocks the reabsorption of urate within the renal proximal tubule. It is intended for the treatment of gout after failure of first line therapy and is administered orally at 400mg once daily
A Phase 3 Randomized, Double-Blind, Multicenter, Placebo- Controlled Study to Assess the Efficacy and Safety of Lesinurad Monotherapy Compared to Placebo in Subjects With Gout and an Intolerance or Contraindication to a Xanthine Oxidase Inhibitor
AstraZeneca’s lesinurad (formerly known as RDEA-594) is a selective oral Uric Acid Transporter URAT1 inhibitor currently in Phase III development for the treatment of of gout. The regulatory filings for lesinurad in the US and Europe are expected for the first half of 2014.

Gout (also known as podagra when it involves the big toe), while not life-threatening, is an excruciatingly painful condition caused by a buildup of a waste product in the blood called uric acid, which is normally eliminated from the body through urine. Excess Uric acid crystallizes and get deposited in the joints (usually the big toes), creating symptoms similar to an acute arthritis flare. Gout has seen a recent gradual resurgence as a result of rising obesity rates and poor diet according to a study in the journal Annals of the Rheumatic Diseases.
The current Standard treatment for gout works by inhibiting a protein called xanthine oxidase that helps in the formation of the uric acid. These therapies, some of which have been used for more than 50 years, are not effective in all patients. One is a generic drug called allopurinol that was approved in the U.S. in 1966. The other is febuxostat, marketed by Takeda Pharmaceutical Co. in the U.S. asUloric and by Ipsen SA and others in Europe as Adenuric and approved in the U.S. in 2009.
AstraZeneca’s new product Lesinurad, a selective uric acid re-absorption inhibitor (SURI), tackles gout by blocking a protein called Uric acid trasporter 1 (URAT1) that otherwise would cause the body to reabsorb the uric acid. AstraZeneca acquired lesinurad (aka RDEA-594) as part of its $1.26 billion takeouver of San Diego-based Ardea Biosciences in 2012. RDEA594 is a metabolite of RDEA806, a non-nucleoside reverse transcriptase inhibitor originally developed for HIV.

In top-line results from a Phase III LIGHT study released by AstraZeneca in December 2013 on gout patients who get no benefit from Zyloprim (allopurinol) and febuxostat, lesinurad alone significantly reduced serum levels of uric acid. The company has three other phase III studies ongoing that are testing the use of the drug alongside allopurinol and febuxostat, and these should generate results in the middle of 2014. Analysts at JPMorgan Chase forecast lesinurad alone may have peak sales of $1 billion a year. AstraZeneca also has a second, more potent drug called RDEA3179 to treat elevated levels of uric acid or hyperuricemia. Pfizer’s KUX-1151, licensed from Japan’s Kissei Phmarceuticals, is in early stage development.
Gout is not an automatic success indication of drugmakers. Savient Pharmaceuticals filed for Chapter 11 bankruptcy in October 2013 in the face of a severe cash crisis, having spent hundreds of millions of dollars on its would-be flagship — the gout-fighting drug Krystexxa (pegloticase) — with limited results. Krystexxa (pegloticase), a twice-monthly infusion designed to treat severe chronic gout that doesn’t respond to conventional therapy, was approved by the U.S. Food and Drug Administration in September 2010. Crealta Pharmaceuticals acquired Savient for $120.4 million in December 2013.
Lesinurad
RDEA-594
2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]sulfanyl}acetic acid
CAS number: 878672-00-5 (Lesinurad), 1151516-14-1 (Lesinurad sodium)
Mechanism of Action:once-daily inhibitor of URAT1, a transporter in the kidney that regulates uric acid excretion from the body
US patents:US8242154 , US8173690, US808448
Indication: Gout
Developmental Status: Phase III (US, UK, EU)
Originator: Ardea Biosciences (Acquired by AstraZeneca for $1.26 billion in 2012)
Developer: AstraZeneca
…………………………
http://www.google.co.in/patents/US8242154
Example 8 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)-N-(2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume, to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 mins and the organic layer removed. The aqueous layer was cooled to 0° C. and acidified by treatment with HCl (1N) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3×) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
Example 102 Methyl 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate


Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0° C., and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%).

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).

A solution of 1-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropylnaphthalene (3.1 g, 73%).

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of 1-amino-4-cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) at 0° C. The reaction mixture was stirred for 5 min at 0° C. and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
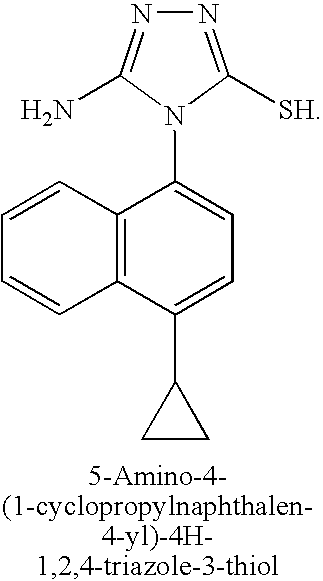
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanatonaphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50° C. for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.0 g, 49%).

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 mins to a suspension of 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
Example 104 Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate

Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 mins to a solution of 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10° C. The mixture was stirred at 10° C. for a further 10 mins. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 103 2-(5-Bromo-4-(1-cyclopropylnapthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 mins to a solution of methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (prepared as described in example 1 above; 1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0° C. The mixture was stirred at 0° C. for a further 45 mins and then neutralized to pH 7 by the addition of 0.5N HCl solution at 0° C. The resulting mixture was concentrated in vacuo to ⅕th of its original volume, then diluted with water (˜20 mL) and acidified to pH 2-3 by the addition of 0.5N HCl to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with DCM is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (1.02 g, 93%).
 |
1H NMR (400 MHz, DMSO-d6) δ ppm 0.84-0.91 (m, 2 H) 1.12-1.19 (m, 2 H) 2.54-2.61 (m, 1 H) 3.99 (d, J = 1.45 Hz, 2 H) 7.16 (d, J = 7.88 Hz, 1 H) 7.44 (d, J = 7.46 Hz, 1 H) 7.59-7.70 (m, 2 H) 7.75 (td, J = 7.62, 1.14 Hz, 1 H) 8.59 (d, J = 8.50 Hz, 1 H) 12.94 (br. s., 1 H) | Mass found: 404.5 (M + 1) | B |
……
POLYMORPHS AND SYNTHESIS
Described herein are various polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate which decreases uric acid levels, (see for example US patent publication 2009/0197825, US patent publication 2010/0056464 and US patent publication 2010/0056465). Details of clinical studies involving sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate have been described in International patent application
PCT/US2010/052958.
Polymorph Form A
In one embodiment, sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H- l,2,4-triazol-3-ylthio)acetate polymorph Form A exhibits an x-ray powder diffraction pattern characterized by the diffraction pattern summarized in Table 1 A or Table IB. In some embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4- cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 3 peaks of (±0.1°2Θ) of Table 1A or IB. In certain embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 4 peaks of (±0.1°2Θ) of Table 1A or IB, at least 5 peaks of (±0.1°2Θ) of Table 1A or IB, at least 6 peaks of (±0.1°2Θ) of Table 1A or IB, at least 8 peaks of
(±0. Γ2Θ) of Table 1A or IB, at least 10 peaks of (±0. Γ2Θ) of Table 1A, at least 15 peaks of (±0. Γ2Θ) of Table 1A, at least 20 peaks of (±0. Γ2Θ) of Table 1A, at least 25 peaks of (±0.1 °2Θ) of Table 1A, or at least 30 peaks of (±0.1 °2Θ) of Table 1A.


Examples
I Preparation of compounds
Example 1: Preparation of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate
Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetate was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.

[00103] Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 min to a solution of 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H- l,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10 °C. The mixture was stirred at 10 °C for a further 10 min. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 2: Preparation of 2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol- 3-ylthio)acetic acid
2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetic acid was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.
[00104] Route i:

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)-N- (2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures, see US 2009/0197825; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 min and the organic layer removed. The aqueous layer was cooled to 0 °C and acidified by treatment with HCl (IN) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3x) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5- bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
[00105] Route ii:

STEP A: 1-Cyclopropylnaphthalene

Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [l,3-bis(diphenylphosphino)propane] dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0 °C, and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%). ] STEP B: l-Cyclopropyl-4-nitronaphthalene

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0 °C. The reaction mixture was stirred at 0 °C for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).
[00108] STEP C: l-Amino-4-cyclopropylnaphthalene

A solution of l-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-amino-4-cyclopropylnaphthalene (3.1 g, 73%).
STEP D: l-Cyclopropyl-4-isothiocvanatonaphthalene

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of l-amino-4- cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in
dichloromethane (50 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield l-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%>).
[00110] STEP E: 5-Amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol

A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), l-cyclopropyl-4- isothiocyanato naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50 °C for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50 °C for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol (2.0 g, 49%). [00111] STEP F: Methyl 2-(5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- -3 -ylthio)acetate

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 min to a suspension of 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room
temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the presence of P2O5 to yield methyl 2-(5- amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).
[00112] STEP G: Methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3 -ylthio)acetate

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room
temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
[00113] STEP H: 2-(5-Bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- )acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 min to a solution of methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3-ylthio)acetate (1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0 °C. The mixture was stirred at 0 °C for a further 45 min and then neutralized to pH 7 by the addition of 0.5N HC1 solution at 0 °C. The resulting mixture was concentrated in vacuo to l/5th of its original volume, then diluted with water (~20 mL) and acidified to pH 2-3 by the addition of 0.5N HC1 to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with dichloromethane is recommended.) The tan solid was
collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the
presence of P2O5 to yield 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- ylthio)acetic acid (1.02 g, 93%).
………………………….
EXAMPLES
The following experiments are provided only by way of example, and should not be understood as limiting the scope of the invention.
COMPOUNDS OF THE INVENTION 2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method A)

1-Cyclopropyl-naphthalene
Cyclopropylmagnesium bromide (150 mL, 0.5 M in tetrahydrofuran) was slowly added to a solution of 1-bromo-naphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloronickel(II) in tetrahydrofuran (10 mL) stirred at 0° C. The reaction mixture was stirred at room temperature for 16 hours and the solvent was evaporated under reduced pressure. EtOAc and ammonium chloride in water were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-naphthalene (6.4 g, 76%).
1-Cyclopropyl-4-nitro-naphthalene
Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropyl-naphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then was slowly poured into ice. Water was added, followed by EtOAc. After extraction, the organic layer was washed with a 1% aqueous solution of NaOH, then washed with water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitro-naphthalene (5.2 g, 64%).
1-Amino-4-cyclopropyl-naphthalene
A solution of 1-cyclopropyl-4-nitro-naphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, then filtered over celite. The solvent was evaporated, and the residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropyl-naphthalene (3.1 g, 73%).
1-Cyclopropyl-4-isothiocyanato-naphthalene
Thiophosgene (1.1 g, 9.7 mmol) was added to a solution of 1-amino-4-cyclopropyl-naphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) stirred at 0° C. The reaction mixture was stirred for 5 min at this temperature, then a 1% solution of HCl in water was added and the organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent was evaporated under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanato-naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was evaporated, toluene was added, and the solvent was evaporated again. A 2.0 M aqueous solution of sodium hydroxide (30 mL) was added and the reaction mixture was heated at 50° C. for 60 hours. The reaction mixture was filtered, and the filtrate was neutralized with a 2.0 M aqueous solution of HCl. New filtration, then evaporation of solvent and purification of the residue by silica gel chromatography to yield 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (2.0 g, 49%).
2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)Acetamide
In a solution of 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (708 mg, 2.5 mmol), K2CO3 (380 mg, 2.5 mmol) in DMF (20 mL) was added 2-chloro-N-(2-chloro-4-sulfamoylphenyl)acetamide (710 mg, 2.5 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction, the solvent was evaporated. The residue was purified by silica gel chromatography to yield 2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (1.26 g, 95%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide
Dichloroacetic acid (180 uL, 2.2 mmol) was added to a suspension of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (0.59 g, 1.1 mmol), sodium nitrite (1.5 g, 22 mmol) and BTEABr (0.91 g, 3.3 mmol) in dibromomethane (30 mL). The reaction mixture was stirred at room temperature for 4 hours, then extracted with dichloromethane and sodium bicarbonate in water. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (224 mg, 31%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method B)

2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole | 2.24 | g | 282.36 | 7.9 |
| methyl chloroacetate | 0.73 | ml | 108.52 | 8.3 (1.05 eq) |
| potassium carbonate | 1.21 | g | 138.21 | 8.7 (1.1 eq) |
| dimethylformamide | 40 | ml | (5 mL/mmol) | |
Procedure:
To a suspension of thiotriazole and potassium carbonate in DMF was added methyl chloroacetate dropwise at room temperature for 5 min. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2.24 g (80%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | ||
| thiotriazole L10183-58 | 709 | mg | 354.43 | 2.0 | |
| bromoform | 10 | ml | (5 ml/mmol) | ||
| sodium nitrite | 2.76 | g | 69.00 | 40 | (20 eq) |
| benzyltriethylammonium | 1.63 | g | 272.24 | 6.0 | (3 eq) |
| bromide | |||||
| dichloroacetic acid | 0.33 | ml | 128.94 | 4.0 | (2 eq) |
Procedure:
To a solution of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester and benzyltriethylammonium chloride in bromoform was added sodium nitrite. To the mixture was added dichloroacetic acid and the reaction mixture was stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel that was packed with CH2Cl2. The column was first eluted with CH2Cl2 until all CHBr3 eluted, and was then eluted with acetone/CH2Cl2 (5:95) to give 713 mg (85%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole methyl ester | 1.14 | g | 418.31 | 2.7 |
| tetrahydrofuran | 10 | ml | (~3 ml/mmol) | |
| ethanol | 10 | ml | (~3 ml/mmol) | |
| water | 10 | ml | (~3 ml/mmol) | |
| lithium hydroxide | 98 | mg | 23.95 | 4.1 (1.5 eq) |
Procedure:
To a solution of 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester, in a mixture of THF and EtOH at 0° C., was added a solution of LiOH in H2O dropwise over 5 min. The reaction was complete after stirring at 0° C. for an additional 45 min. The reaction was neutralized to pH 7 by the addition of 0.5 N HCl solution at 0° C., and the resulting mixture was concentrated in vacuo to ⅕th of its original volume. The mixture was diluted with H2O (˜20 mL) and acidified to pH 2-3 by the addition of 0.5 N HCl to produce sticky solid. (If the product comes out as an oil during acidification, extraction with CH2Cl2 is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 1.02 g (93%) of the title compound.
REF:
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8242154 B2 ;Also published as US20100056465, US20130040907;Original Assignee: Ardea Biosciences, Inc
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8173690 B2;Also published as US20100056464;Original Assignee: Ardea Biosciences, Inc
Barry D. Quart, Jean-Luc Girardet, Esmir Gunic, Li-Tain Yeh;Compounds and compositions and methods of use;US patent number US8084483 B2; Also published as CA2706858A1, CA2706858C, CN101918377A, CN102643241A, CN103058944A, EP2217577A2, EP2217577A4, US8283369, US8357713, US8546437, US20090197825, US20110268801, US20110293719, US20120164222, US20140005136, WO2009070740A2, WO2009070740A3;Original Assignee:Ardea Biosciences, Inc.
Gunic, Esmir; Galvin, Gabriel;Manufacture of 2-[5-bromo-4-(cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio]acetic acid and related compounds;PCT Int. Appl., WO2014008295 A1
Zamansky, Irina et al;Process for preparation of polymorphic, crystalline, and mesophase forms of 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]acetic acid sodium salt; PCT Int. Appl., WO2011085009
Gunic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels; U.S. Pat. Appl. Publ., 20100056465
unic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels;U.S. Pat. Appl. Publ., 20100056464
Quart, Barry D. et al;Preparation of azole carboxylates as modulators of blood uric acid levels;PCT Int. Appl., 2009070740, 04 Jun 2009
Girardet, Jean-Luc et al;Preparation of S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase;PCT Int. Appl., WO2006026356
US20100056465 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
US20100056542 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
WO2009070740A2 * Nov 26, 2008 Jun 4, 2009 Ardea Biosciences Inc Novel compounds and compositions and methods of use
WO2011085009A2 * Jan 5, 2011 Jul 14, 2011 Ardea Biosciences, Inc. Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof
| WO2011159732A1 * | Jun 14, 2011 | Dec 22, 2011 | Ardea Biosciences,Inc. | Treatment of gout and hyperuricemia |
| WO2012092395A2 * | Dec 28, 2011 | Jul 5, 2012 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio) acetic acid and uses thereof |
| EP2560642A2 * | Mar 29, 2011 | Feb 27, 2013 | Ardea Biosciences, Inc. | Treatment of gout |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US20100056465 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20100056542 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| WO2011085009A2 * | Jan 5, 2011 | Jul 14, 2011 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof |
| US8283369 | 30 Jun 2011 | 9 Oct 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | 30 Jun 2011 | 22 Jan 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8546437 | 30 Jun 2011 | 1 Oct 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8633232 * | 4 May 2012 | 21 Jan 2014 | Ardea Biosciences, Inc. | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20130040907 * | 4 May 2012 | 14 Feb 2013 | Ardea Biosciences Inc. | Compounds, Compositions and Methods of Using Same for Modulating Uric Acid Levels |
| WO2006026356A2 * | Aug 25, 2005 | Mar 9, 2006 | La Rosa Martha De | S-TRIAZOLYL α-MERCAPTOACETANILDES AS INHIBITORS OF HIV REVERSE TRANSCRIPTASE |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| US20090197825 * | Nov 26, 2008 | Aug 6, 2009 | Ardea Biosciences, Inc. | Novel compounds and compositions and methods of use |
| US7947721 | Aug 25, 2005 | May 24, 2011 | Ardes Biosciences Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8084483 | Nov 26, 2008 | Dec 27, 2011 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8106205 | Feb 2, 2010 | Jan 31, 2012 | Ardea Biosciences, Inc. | N[S(4-aryl-triazol-3-yl)α-mercaptoacetyl]p-amino benzoic acids as HIV reverse transcriptase inhibitors |
| US8252828 | Jun 30, 2011 | Aug 28, 2012 | Ardea Biosciences, Inc. | S-triazolyl α-mercapto acetanilides as inhibitors of HIV reverse transcriptase |
| US8283369 | Jun 30, 2011 | Oct 9, 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | Jun 30, 2011 | Jan 22, 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8372807 | May 20, 2010 | Feb 12, 2013 | Ardea Biosciences, Inc. | Methods of modulating uric acid levels |
| US8481581 | Jul 18, 2012 | Jul 9, 2013 | Ardea Biosciences, Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8524754 | Jan 5, 2011 | Sep 3, 2013 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio) acetate, and uses thereof |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US8546437 | Jun 30, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |

Sorry, the comment form is closed at this time.