












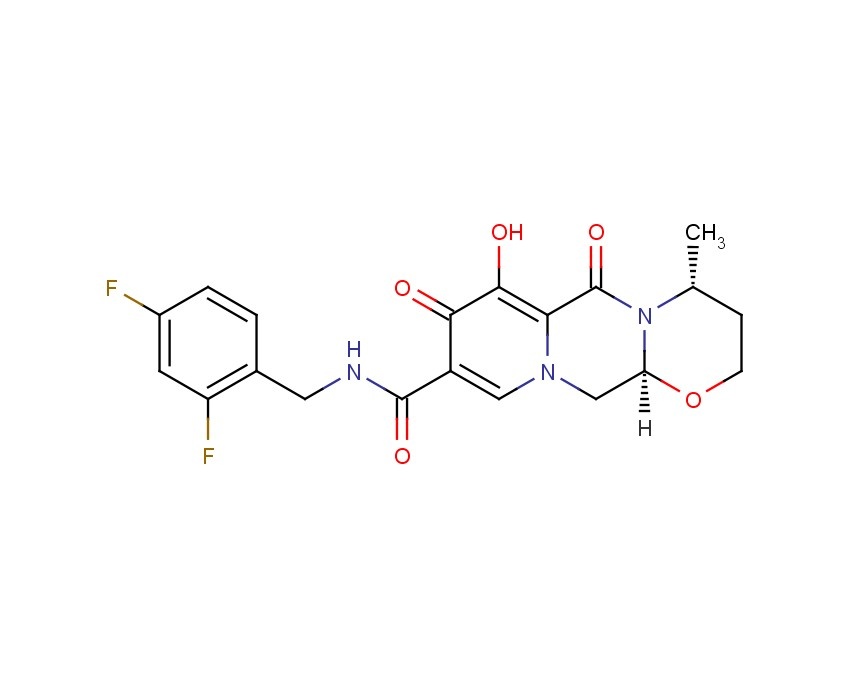
Dolutegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.

Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
……………………………………
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
-
Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

-
This document describes the following reaction formula as a method of producing compound (I).


-
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………………………………
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
………………………………………………………
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

………………………
SYNTHESIS
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
………………..
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
-
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
-
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
-
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
-
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
-
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
-
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
-
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
-
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
-
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
-
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
-
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
-
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2
-

-
Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
……………..

ENTRY 68
………………………….
WO 2010068262
…………………………
WO 2010068253
…………………………………
WO 2011119566
…………………………..
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- “U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay”. Reuters. 12 August 2013. Retrieved 13 February 2013.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- “ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV”. Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- “Dolutegravir Prescribing Information”. Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). “Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.”. The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- Jump up^ http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). “Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection”. New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. “SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA – See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf”. Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). “Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.”. The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
-
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds -
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html

Sorry, the comment form is closed at this time.